Angeborene Herzfehler (Band 1)
Die Informationen auf dieser Seite finden Sie in Band 20 einer eBook-Reihe der Patienten-Akademie.
Hier bekommen Sie dieses eBook in verschiedenen Formaten:
Band 1:
- padBook (für iPad und epub3-fähige eBook-Reader)
- phoneBook (für smartPhones)
- Paperwhite (für Kindle Paperwhite)
Leseprobe Band 1
Viele der Herzfehler lassen sich in ihrer Entstehung verstehen, wenn man weiß, wie sich das Herz und der Kreislauf mit seinen Gefäßen im Mutterleib entwickeln, d.h. wenn man die „embryonale Herzentwicklung“ kennt.
Eine detaillierte Schilderung dieses sehr komplexen Vorgangs würde den Rahmen dieses eBooks sprengen. Wenn Sie sich dennoch dafür interessieren empfehle ich ein eBook, daß ich zu diesem Thema für Ärzte geschrieben habe und das Sie bekommen können, wenn Sie im Corobuch auf Band 26 und 27 klicken.
Wie gesagt, es ist kompliziert, aber für ein grundlegendes Verständnis wichtig.
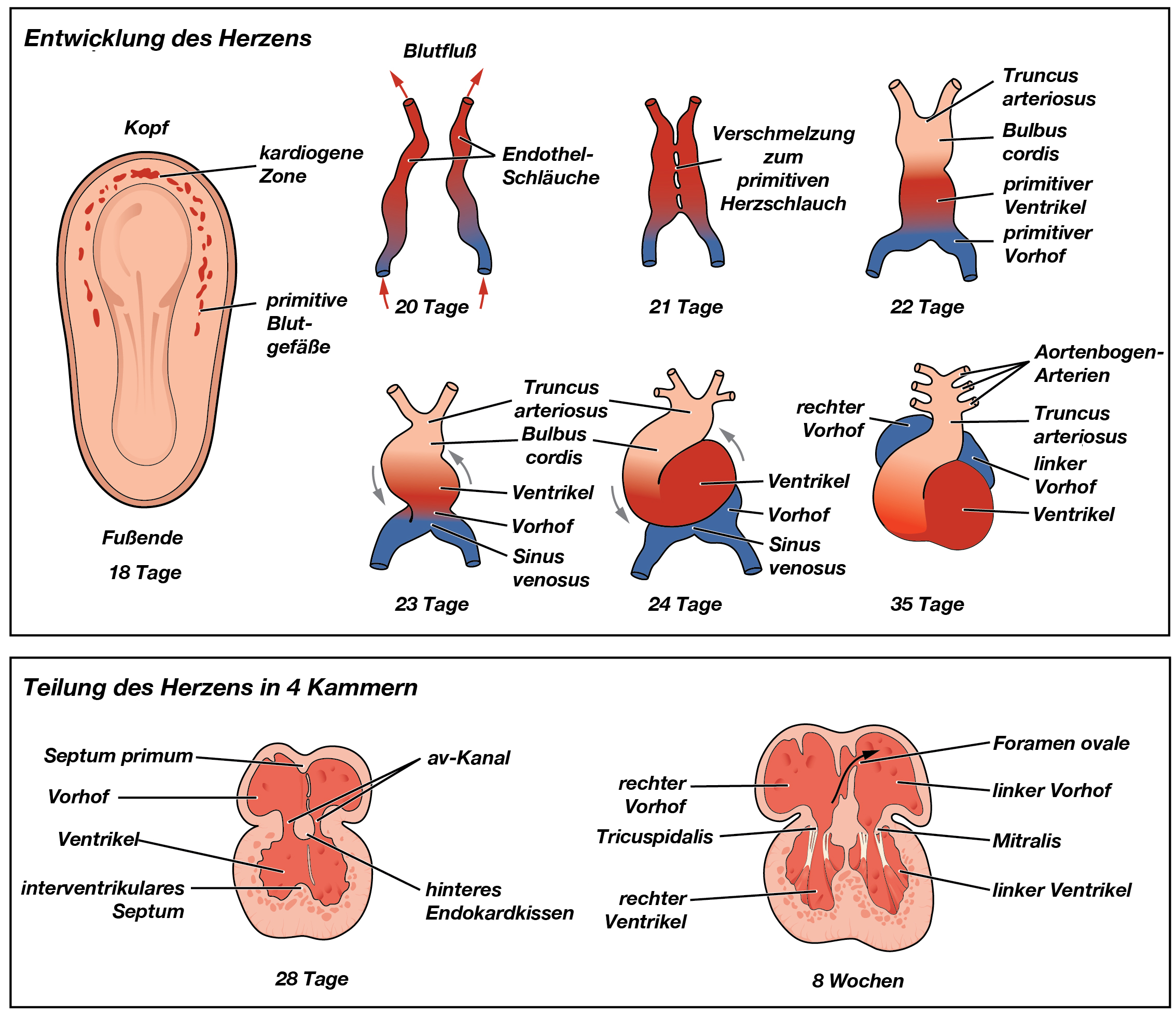
Entwicklung des Herzens
Eine detaillierte Schilderung dieses sehr komplexen Vorgangs würde den Rahmen dieses eBooks sprengen.
Wenn Sie sich dennoch dafür interessieren empfehle ich ein eBook, daß ich zu diesem Thema für Ärzte geschrieben habe und das Sie bekommen können, wenn Sie hier klicken.
Wie gesagt, es ist kompliziert, aber für ein grundlegendes Verständnis wichtig. Nur um es kurz zusammenzufassen (Abb. 1):
Entwicklung des Herzschlauches
 |
| Abb. 1 |
Das Herz entsteht aus 2 Schläuchen, die mit Gefäßinnenhaut-Zellen (Endothel) ausgekleidet sind.
Diese beiden Schläuche verschmelzen etwa in der 3. Schwangerschaftswoche zu einem einzigen Schlauch, dem sog. Herzschlauch. Durch unterschiedliches Längen- und Dickenwachstum bestimmter Stellen dieses Schlauches dreht, knickt und faltet sich das primitive Herz zur Herzschleife. Dieser Vorgang ist etwa zur 35. Schwangerschaftswoche abgeschlossen, wobei das Innere des Herzschlauches bis hierhin nur aus 1 Hohlraum besteht.
Teilung des primitiven Herzens
In der Mitte des späteren Herzens befindet sich ein ganz spezielles Gewebe, die sog. Endokardkissen, von denen es insgesamt 4 gibt (vorderes, hinteres und 2 seitliche Kissen).
Durch das Wachstum dieser Kissen in verschiedene Richtungen (nach oben, nach unten, nach rechts und nach links) wird der Innenraum des primitiven Herzens in 4 Kammern unterteilt.
Dadurch, daß diese Strukturen teilweise aus verschiedenen Richtungen aufeinander zuwachsen und miteinander verschmelzen entstehen Trennwände zwischen den Vor- und Hauptkammern. Besondere Erwähnung verdient hierbei die Trennwand zwischen den beiden Vorkammern (Vorhofseptum):
Es entwickelt sich nämlich nicht nur 1, sondern gleich 2 Trennwände (Septum primum und Septum secundum), die jeweils ein Öffnung haben (Ostium primum und Ostium secundum) (Genaueres erfahren Sie im Kapitel über den Vorhofseptumdefekt in Band 2).
Diese beiden Löcher liegen nicht genau übereinander, sondern sind gegeneinander etwas versetzt.
Dadurch, daß beide Trennwände beweglich sind entsteht eine Art Ventil zwischen beiden Vorkammern, das es ermöglicht, daß Blut von der rechten in die linke Vorkammer, nicht aber umgekehrt fließt. Dieser Mechanismus ist für den Blutkreislauf des Kindes im Mutterleib, wenn die Lungen noch keine Luft enthalten, lebenswichtig.
Entwicklung der Aorta
Die Entwicklung der Hauptschlagader, der Aorta, verläuft relativ komplex:
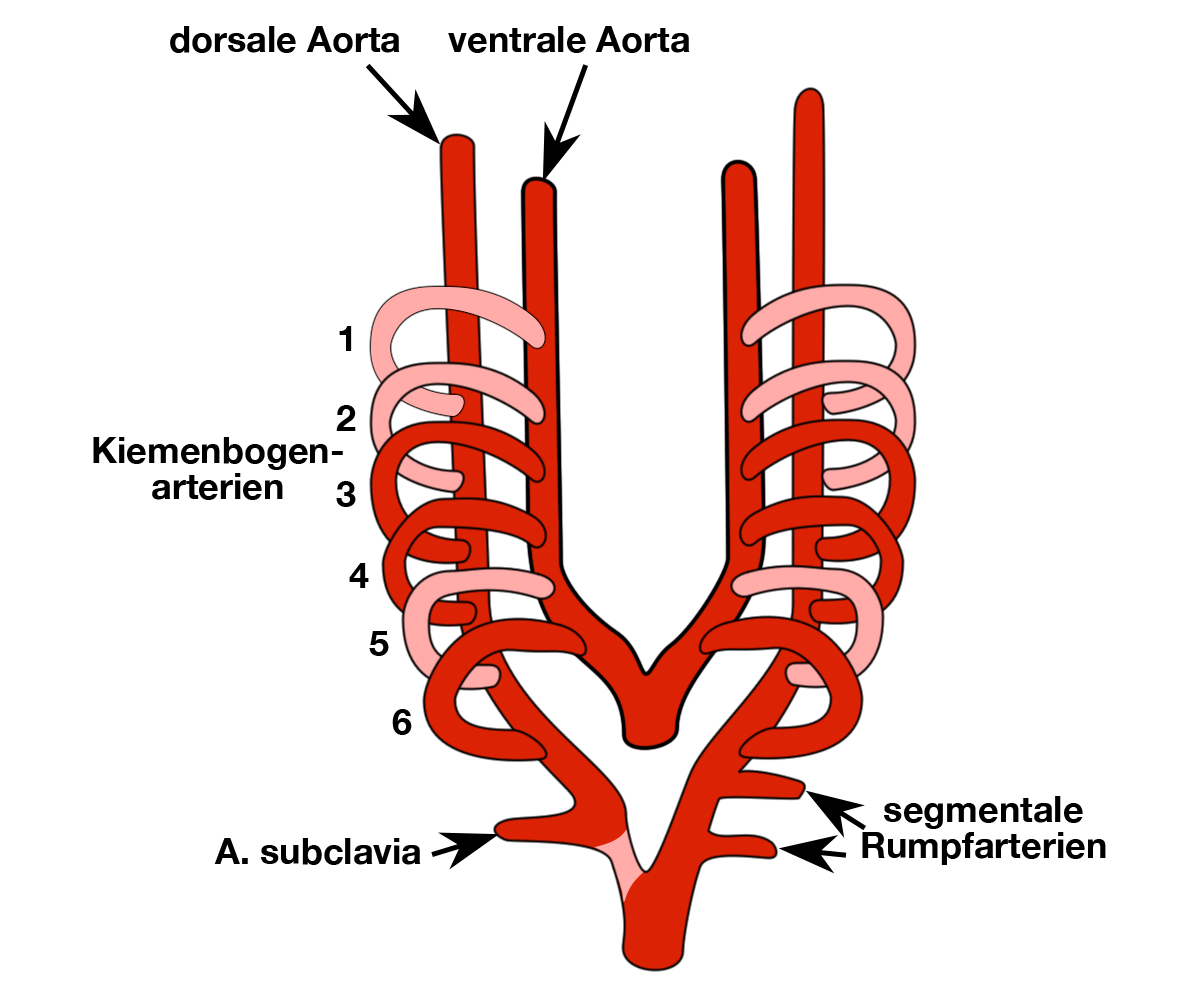
 |
| Abb. 2 |
Die Aorta entwickelt sich ab der 5. Woche der embryonalen Entwicklung, deren Vorstufen haben sich aber schon vorher entwickelt:
Beteiligt sind der Truncus arteriosus, der nach kopfwärts in den Aortensack übergeht und die hiervon unabhängig entstandenen hinteren (= dorsalen) Aorten (Abb. 2).
Diese hinteren dorsalen Aorten entwickelt sich unabhängig von der Herzanlage aus 2 primitiven Gefäßschläuchen, die im rückenwärtigen Teil des Embryos paarig angelegt sind.
Gleichzeitig bildet der Aortensack als Verlängerung des Truncus arteriosus in seinem oberen Anteil 2 Aussackungen (Hörner) aus.
Mit zunehmendem Wachstum entwickeln sich aus diesen Hörnern auf beiden Seiten die beiden vorderen (= ventralen) Aorten (Abb. 2).
Sowohl die beiden dorsalen als auch die beiden ventralen Aorten wachsen im Verlauf nach oben (kopfwärts) fort und verschmelzen am Ende der 4. Woche.
 |
| Abb. 3 |
| Ende der 4. Woche: Die in rosa eingezeichneten Kiemenbogenarterien bilden sich im Verlauf zurück. |
Von beiden ventralen Aorten ausgehend bilden sich insgesamt 6 Paare von Kiemenbogenarterien, die aber nie gleichzeitig vorhanden sind, weil sie sich nacheinander entwickeln.
Diese Kiemenbogenarterien verbinden sich ab der 5. Woche mit den beiden dorsalen Aorten (Abb. 3).
Damit sind beide dorsalen Aorten über die Kiemenbogenarterien mit den ventralen Aorten verbunden.
Die beiden dorsalen Aorten verschmelzen im weiteren Verlauf der Entwicklung unterhalb der Mündung der 6. Kiemenbogenarterie zur Aorta descendens (siehe Abb. 2).
Diese Vereinigung schreitet von unten nach oben immer weiter fort.
Im weiteren Verlauf der Entwicklung kommt es zu erheblichen Umbildungen dieser Strukturen.
Indem sich ein Teil der Kiemenbogenarterien weiter entwickelt und ein anderer Teil verkümmert (siehe Abb. 3) bilden sich die Aorta und die aus ihr entspringenden Gefäße zu ihrer endgültigen Gestalt.
Wichtig ist in diesem Zusammenhang die 6. Kiemenbogenarterie, aus der sich der sog. Ductus arteriosus BOTALLI entwickelt:
Der Ductus ist während des Lebens des Kindes im Mutterleib eine wichtige Verbindung zwischen dem Lungen- und dem Organkreislauf.
Entwicklung der Herzklappen
Es gibt im Herzen 4 Klappen:
- 2 Klappen befinden sich am Ausgang des rechten bzw. linken Ventrikels und leiten das Blut in die Lungenschlagader (Pulmonalklappe) bzw. in die Aorta. Bei diesen Klappen handelt es sich um die sog. Taschenklappen.
- Die beiden anderen Klappen befinden sich zwischen den Vorhöfen und den Ventrikeln (Tricuspidalklappe zwischen rechtem Vorhof und rechtem Ventrikel bzw. Mitralklappe zwischen linkem Vorhof und linkem Ventrikel). Diese Klappen sind wie Fallschirme gestaltet, d.h. sie bestehen aus einem Segel, dessen Ränder mit Sehnenfäden verbunden sind, die wiederum im Herzmuskel der Ventrikel verankert sind. Diese Klappen bezeichnet man als Segel- oder, weil sie zwischen den Vorhöfen (a) und den Ventrikeln (v) liegen auch als av-Klappen.
Die Entwicklung dieser beiden Klappentypen ist unterschiedlich und verläuft unter Beteiligung der Endokardkissen.
Dabei entstehen die verschiedenen Segel und Taschen der einzelnen Klappe aus unterschiedlichen Endokardkissen entstehen. Auf die Entwicklung der jeweiligen Klappen wird nachfolgend bei den jeweiligen angeborenen Fehlentwicklungen eingegangen.
Entwicklung des Kreislaufs
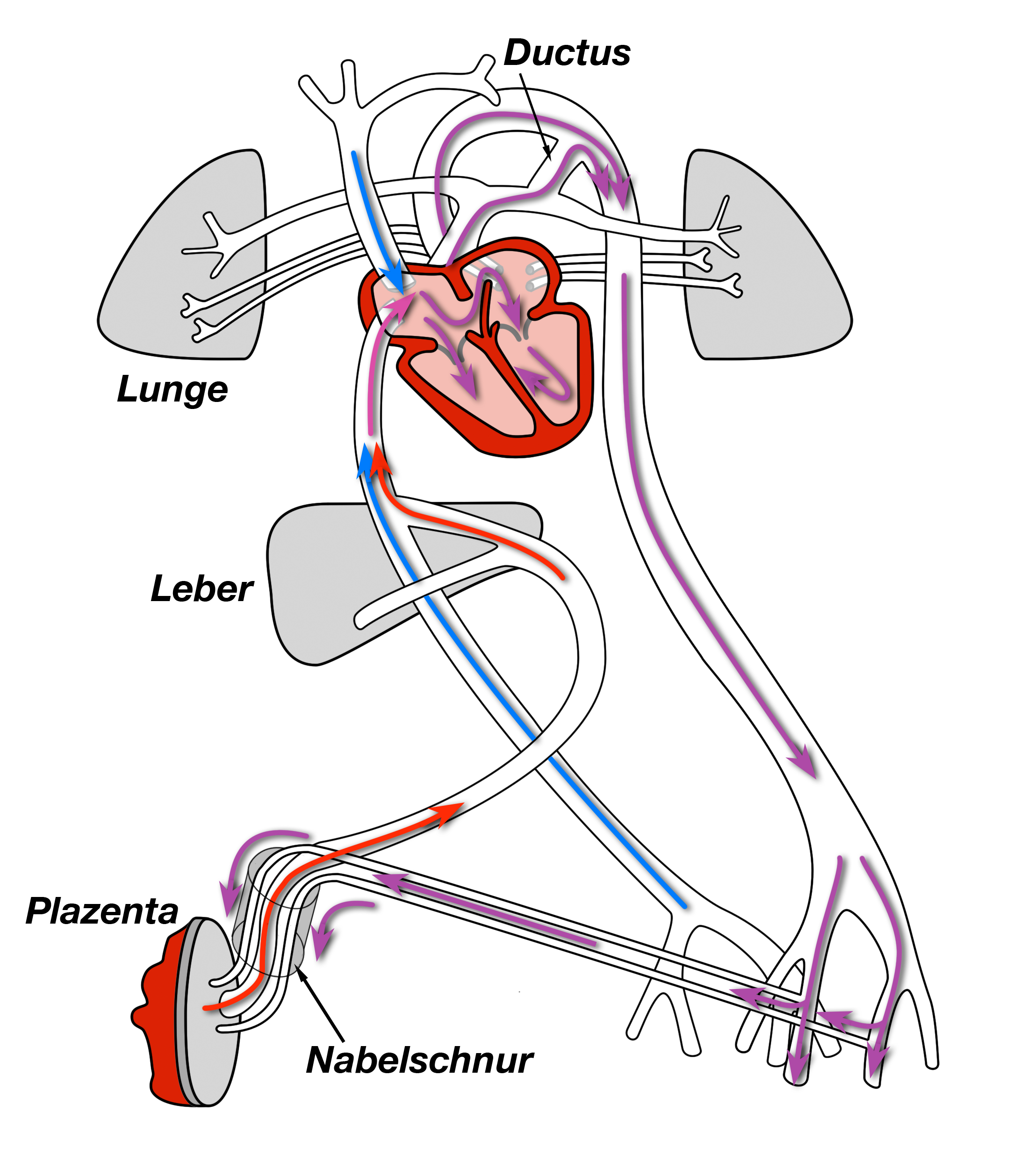 |
| Abb. 4 |
| Ende der 4. Woche: Die in rosa eingezeichneten Kiemenbogenarterien bilden sich im Verlauf zurück. |
Solange sich das Kind im Mutterleib befindet haben seine Lungen keine Funktion, denn es gibt ja keine Luft zum Atmen. Daher ist der Kreislauf im Mutterleib so gestaltet, daß das Blut an den Lungen vorbei strömt (Abb. 4):
Die Aufgabe der Lunge übernimmt beim ungeborenen Kind der Mutterkuchen (= Plazenta).
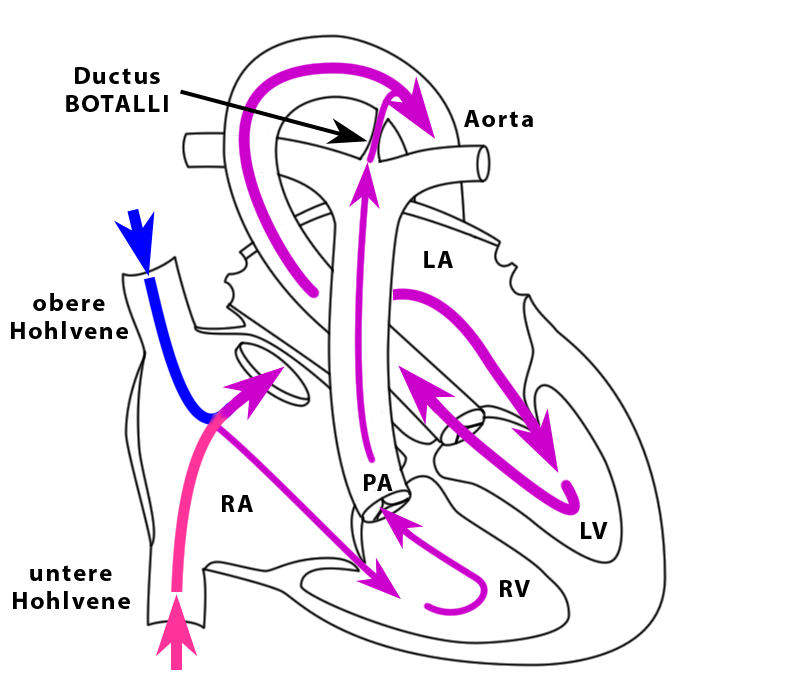 |
| Abb. 5 |
Hier tritt der Blutsauerstoff des mütterlichen Blutes in das Blut des Kindes über. Von der Plazenta aus fließt das sauerstoffreiche Blut durch die Nabelvene in der Nabelschnur des Kindes im Bereich der Leber in die untere Hohlvene des Kindes und von hier aus in den rechten Vorhof (Abb. 5).
Solange die Lungen nicht benötigt werden ist der Flußwiderstand der Lungenschlagader deutlich erhöht.
Dieser hohe Widerstand soll verhindern, daß Blut aus dem rechten Ventrikel in die (noch funktionslosen) Lungen fließt.
Um das Blut aber tatsächlich an der Lunge vorbei zu führen strömt es durch das zu diesem Zeitpunkt noch offene Foramen ovale (= Ostium primum + Ostium secundum) in den linken Vorhof und von hieraus durch den linken Ventrikel und die Aorta in die Organe des Kindes.
Das verbrauchte Blut schließlich kehrt über Verbindungsgefäße zwischen den Beckenvenen, d.i. über die Nabelarterie zurück in die Plazenta, um hier wieder mit Sauerstoff angereichert zu werden.
Durch das Foramen ovale fließt aber nicht das gesamte sauerstoffreiche Blut der Nabelvene, das sauerstoffarme Blut aus der oberen Körperhälfte und derjenige Teil des sauerstoffarmen Blutes, der nicht über die Nabelarterie in die Plazenta geleitet wird. Ein gewisser Anteil dieses Blutes fließt nämlich durch den rechten Vorhof hindurch und gelangt damit über den rechten Ventrikel in die Lungenschlagader.
Damit dieses Blut nicht unnötigerweise durch die funktionslosen, weil luftleeren Lungen strömen muß fließt es über den Ductus arteriosus aus der Lungenschlagader direkt in die Aorta und vermischt sich hier mit dem sauerstoffreichen Blut, das über das Foramen ovale dorthin geflossen ist (siehe Abb. 5).
Unmittelbar nach der Geburt, oft schon mit den 1. Atemzügen des neugeborenen Kindes, meistens aber erst innerhalb weniger Tage bis Wochen, sinkt der bis dahin erhöhte Fließwiderstand in den Lungenschlagadern ab, sodaß es dem Blut von hier an leichter möglich ist durch die Lungen zu fließen. Foramen ovale und der Ductus arteriosus werden nun nicht mehr benötigt und sie verschließen sich wieder.
| Film 1 |
Dies ist zu diesem Zeitpunkt unbedingt notwendig, denn von nun an wird der Sauerstoff in den Lungen des Kindes ins Blut aufgenommen (Film 1).
Die angeborenen Herzfehler entstehen dadurch, daß der oben skizzierte Ablauf bei der Entstehung des Herzens und der Aorta infolge genetischer Veränderungen gestört wird. Die Art der genetischen Störung selber ist in vielen Fällen nicht genau bekannt, deren Auswirkungen in vielen Fällen aber durchaus.
So kommt es beispielsweise durch eine Fehlentwicklung der Vorhofseptum dazu, daß sich deren Löcher nicht verschließen und daß dadurch auch nach der Geburt eine Verbindung zwischen beiden Vorkammern verbleibt (Vorhofseptumdefekt). Durch solche Löcher fließt Blut beispielsweise von der rechten in die linke Vorkammern, was Auswirkungen auf den Sauerstoffgehalt des Blutes im „großen“ Kreislauf und damit auf die Sauerstoffversorgung der Organe hat.
Kommt es zu einer Fehlentwicklung bestimmter Herzklappen entstehen „mißgebildete“ Klappen, die entweder zu eng (= Stenose) oder undicht (= Insuffizienz) sind.
Außer in speziellen Fällen möchte ich bei der nachfolgenden Schilderung einzelner angeborener Herzfehler nicht darauf eingehen, welche Entwicklungsstörung die genaue Ursache des Herzfehlers ist, sondern es dabei belassen, daß es halt eben Störungen der Herzentwicklung sind. Einteilung der angeborenen Herzfehler Um die verschiedenen angeborenen Herzfehler zu unterscheiden betrachtet man solche mit und solche ohne „Shunt“.
„Shunt“ ist dabei die Bezeichnung für einen Kurzschluß zwischen den Kreisläufen. Sie wissen aus den ersten Kapiteln dieser eBook-Reihe, den denen ich über den Kreislauf und die Anatomie des Herzens berichtet habe, daß es 2 Kreisläufe (den „großen“ und den „kleinen“, d.i. ist den arteriellen und den venösen Kreislauf) gibt, die streng voneinander getrennt sind.
Durch bestimmte Entwicklungsstörungen des Herzens und des Kreislaufes kann diese Trennung durch Löcher in Trennwänden oder „falsch angeschlossene und verbundene“ Gefäße aufgehoben werden. Durch solche Defekte kommt es dann zu einer Durchmischung des Blutes beider Kreisläufe. Dies hat u.a. zur Folge, daß sich arterielles, d.i. sauerstoffreiches Blut und venöses, d.i. sauerstoffarmes Blut in unterschiedlichem Ausmaß vermischen, was, wie Sie sich vorstellen können, zu vielen Problemen führen kann.
Also: Lesen Sie zunächst etwas über angeborene Herzfehler ohne Shunt.
Aortenstenose
Beschreibung der Erkrankung
Bei der erworbenen Aortenstenose ist lediglich die Aortenklappe verengt. Unter dem Begriff der angeborenen Aortenstenose werden Verengungen zusammengefaßt, die nicht nur die Klappe selber, sondern auch den unmittelbar unterhalb, d.h. noch im linken Ventrikel befindlichen Bereich und die Aorta oberhalb der Klappe betreffen.
Es ist hilfreich, wenn man die embryonale Entwicklung dieser Herz- und Gefäßteile kennt.
Embryonale Entwicklung
 |
| Abb. 6 |
| Entstehung des primitiven Herzschlauches |
| A: 2 parallel verlaufende Endokardschläuche, die sich |
| B und C: miteinander vereinigen und den primitiven Herzschlauch bilden |
Am 22. Tag der Schwangerschaft vereinigen sich die beiden ursprünglich entstandenen, parallel verlaufenden Endokardschläuche zum sog. Herzschlauch (Abb. 6).
 |
| Abb. 7 |
| Bestandteile des Herzschlauches |
Er gliedert sich in mehrere Abschnitte (Abb. 7), wobei der
- sog. Bulbus cordis für die Entstehung der Aorten- und Pulmonalklappe,
- der Truncus arteriosus für die Entstehung der beginnenden Aorta (siehe im Abschnitt über die Aortenisthmusstenose) und
- der Conus cordis für die Entstehung des unmittelbar unterhalb der Aortenklappe liegenden Ausflußtraktes des linken Ventrikels
zuständig ist.
Die Entwicklung der Aortenklappe erfolgt im Bulbus cordis (Abb. 8).
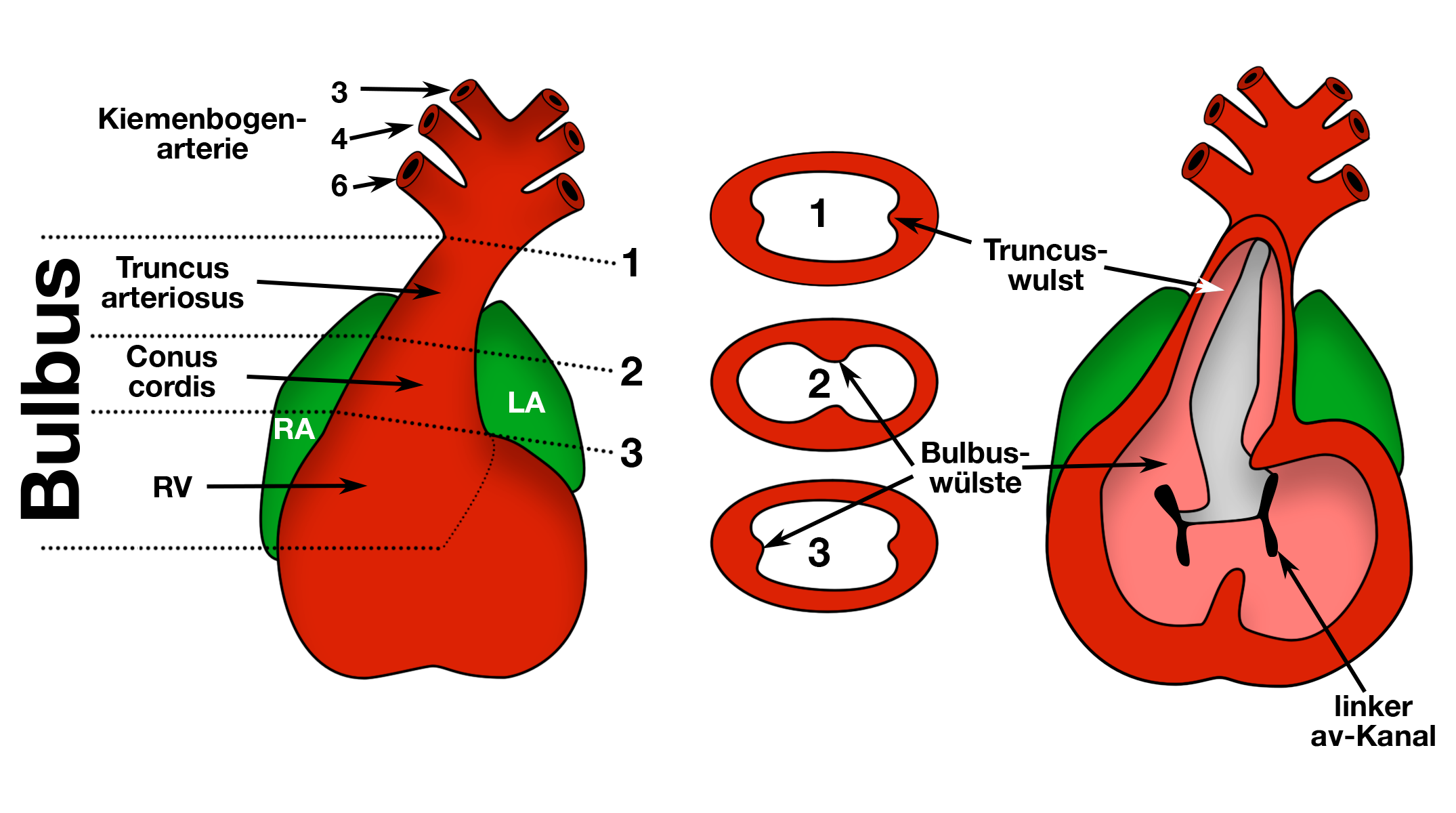 |
|
| Abb. 8 | |
| Außenansicht (links), Innenansicht (rechts) Querschnitte an 3 Ebenen des Bulbus cordis (1, 2 und 3) |
Hier bilden sich 4 Bulbus-Wülste: Der rechte und der linke, der vordere und der hintere Wulst (Abb. 9).
 |
| Abb. 9 |
dorsaler = hinterer, ventraler = vorderer Wulst |
Sie liegen direkt unterhalb von 2 anderen Wülsten, die sich direkt oberhalb im Truncus arteriosus entwickeln und die daher Truncuswülste genannt werden.
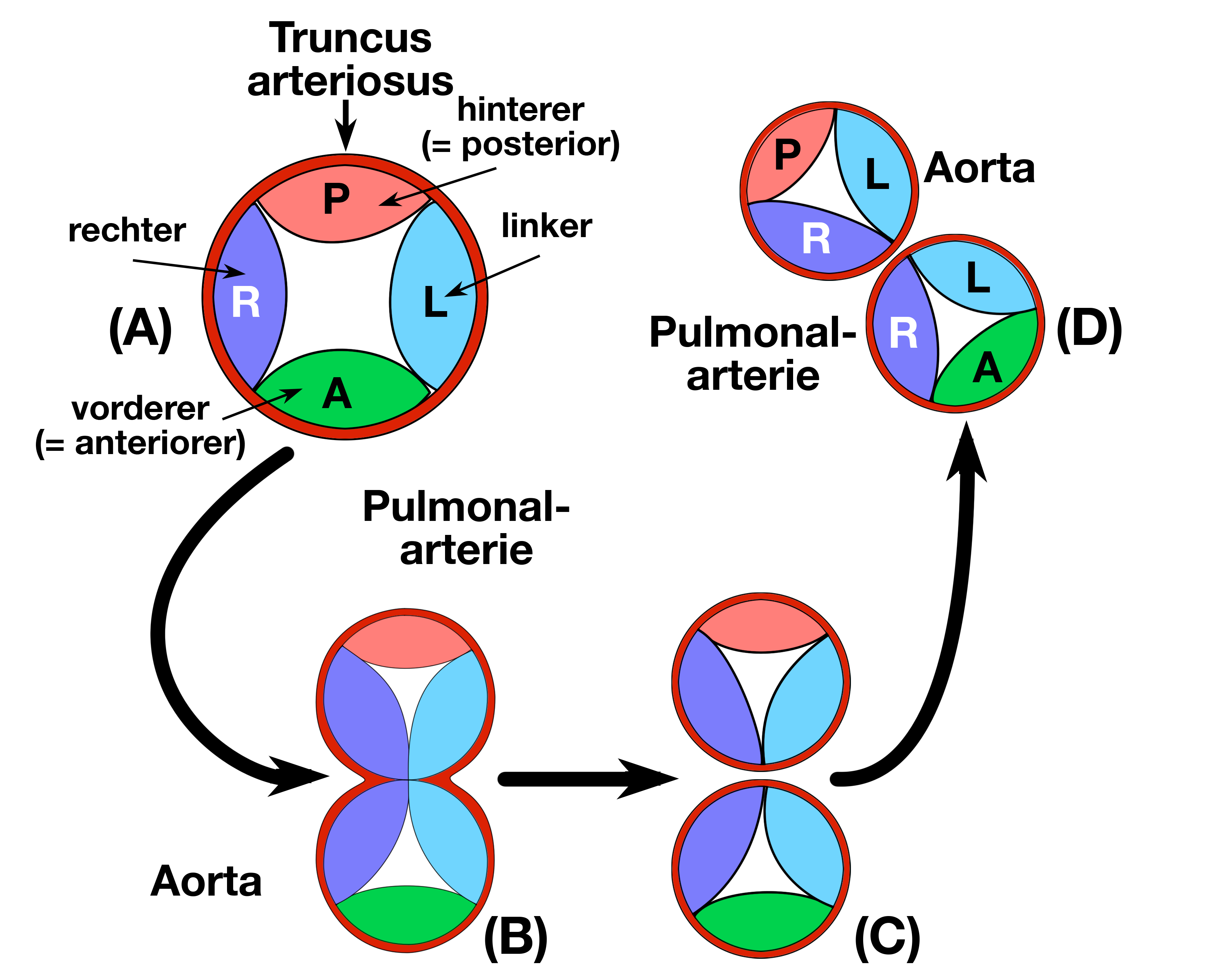 |
| Abb. 10 |
| Drehung der Bulbuswülste A: Ausgangszustand: Truncus arteriosus mit den 4 Bulbuswülsten B: Aufteilung der Truncus arteriosus durch rechten und linken Bulbuswulst C: Trennung von Aorta und Pulmonalarterie D: Drehung von Aorta und Pulmonalarterie (Pulmonalarterie liegt vorne, Aorta hinten) Pulmonalarterie = Lungenschlagader |
Wenn die Truncuswülste wachsen verschmelzen sie miteinander und trennen hierdurch den Truncus arteriosus in 2 Gefäße, aus denen sich später die Aorta und die Lungenschlagader (Arteria pulmonalis) entwickeln. Gleichzeitig wachsen der rechte und linke Bulbuswulst aufeinander zu und auch nach oben in Richtung auf die Truncuswülste, sodaß sie schließlich alle miteinander verwachsen (Abb. 10).
Wenn die Aufteilung des Truncus arteriosus abgeschlossen ist und somit eine aortale und eine pulmonale Öffnung geschaffen wurde verteilen sich die Bulbuswülste um diese beiden Öffnungen wie folgt:
Die aortale Öffnung wird vom hinteren, vom rechten und vom linken Bulbuswulst, die pulmonale Öffnung vom vorderen und ebenfalls von Teilen des rechten und linken Bulbuswulstes gebildet, d.h. jede Öffnung wird nun von 3 Bulbuswülsten begrenzt (siehe Abb. 10).
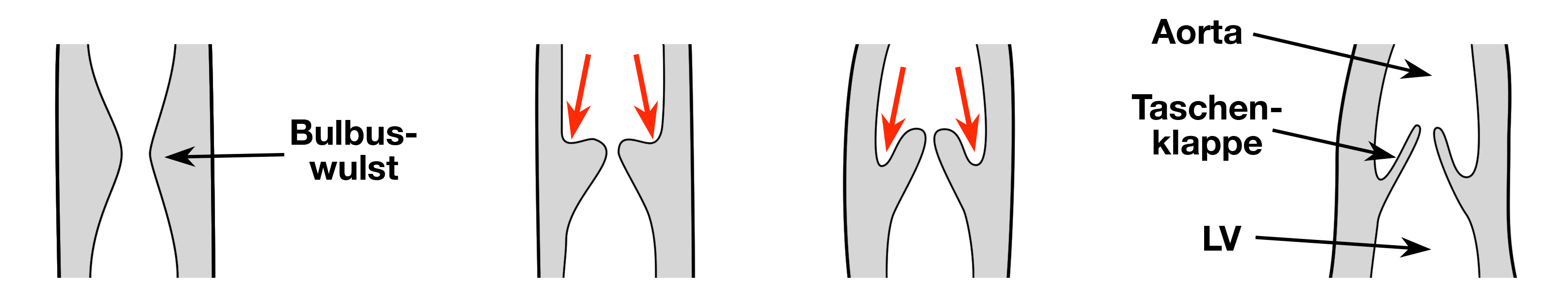 |
| Abb. 11 |
| Entwicklung der Taschenklappen |
Aus diesen Wülsten entwickeln sich in der Folge die Klappentaschen. Die Aorten- (und ebenfalls die Pulmonal-) Klappe entwickeln sich durch ein aktive Wachstum der Bulbuswülste (Abb. 11).
Durch Wachstum, zunehmende Verdünnung dieser Gebilde und unter Einwirkung des Drucks in den Gefäßen entstehen letztlich die dünnhäutigen Taschenklappen. Der Umstand, daß die Aorten- (ebenso wie die Pulmonal-) Klappe aus 3 Teilen der Bulbuswülste entsteht ist der Grund dafür, warum diese Klappen im Normalfall aus jeweils 3 einzelnen Taschenklappen bestehen.
Ursache der Aortenstenosen
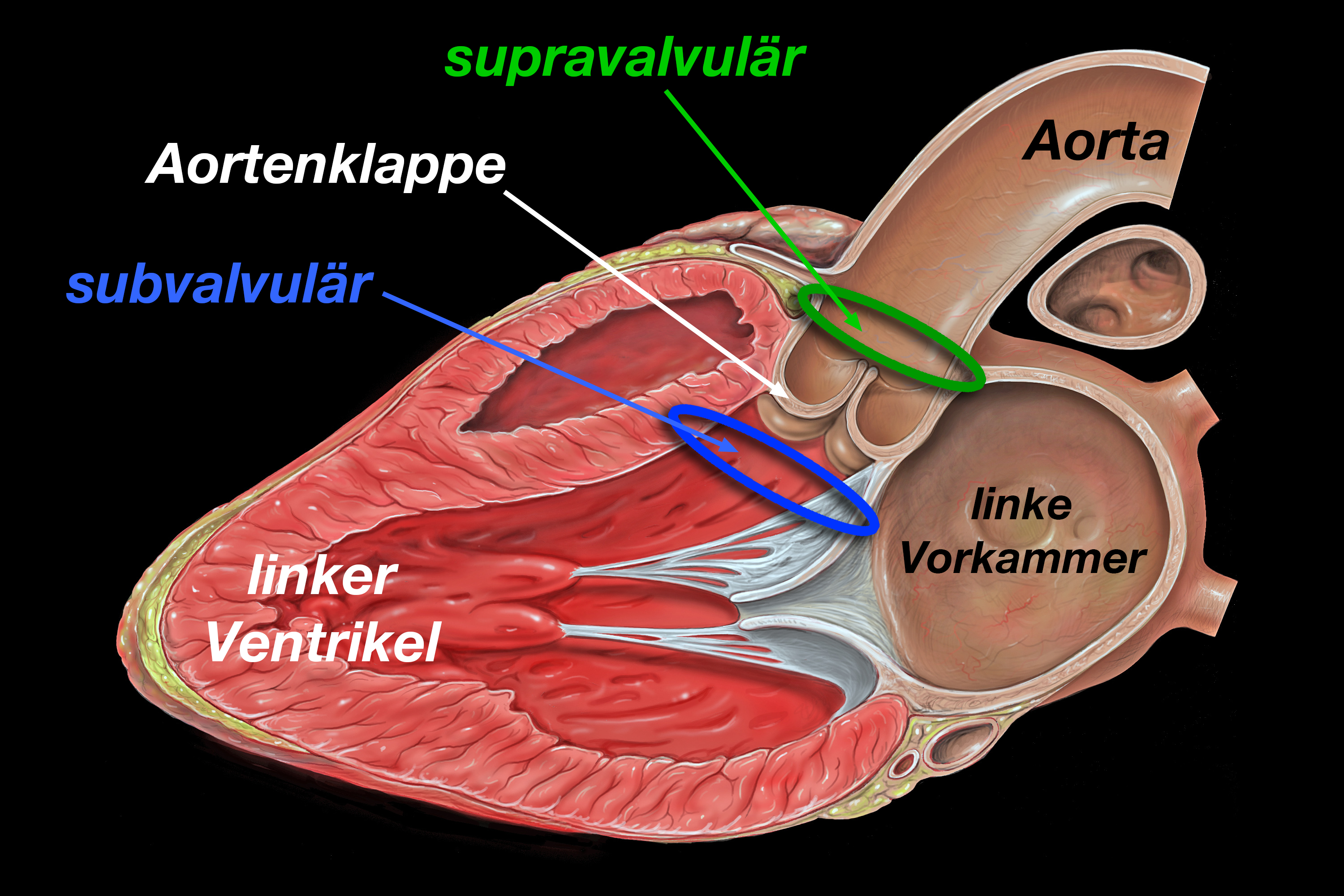 |
| Abb. 12 |
Es gibt verschiedene Arten der angeborenen Aortenstenose, die sich nach dem Sitz der Verengung unterscheiden (Abb. 12):
- Bei der valvulären Form ist die Klappe selber (= Valvula) betroffen. Entweder sind die einzelnen Taschen der Klappe miteinander verklebt oder die Klappe besteht nicht (wie normalerweise) aus 3, sondern nur aus 2 Taschen (Abb. 13).
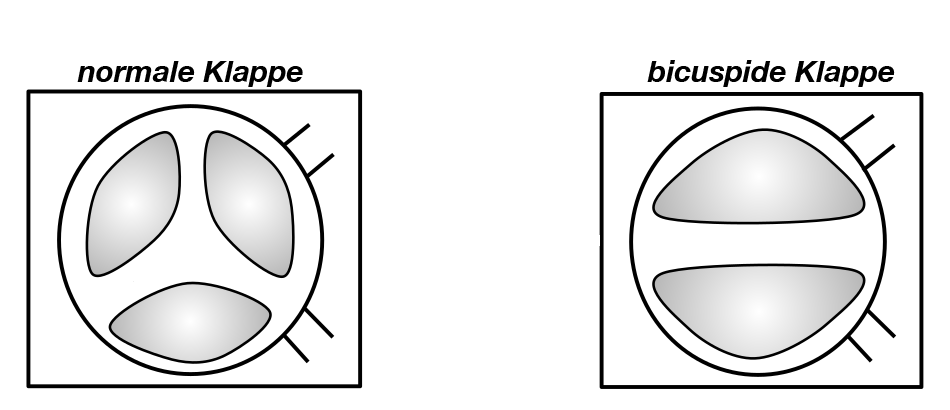
Abb. 13 Links: Gesunde Aortenklappe mit 3 Taschen, rechts: Angeborene Aortenklappenstenose mit nur 2 Taschen Ursache hierfür ist eine Störung in der Anordnung und Drehung der Bulbuswülste.
- Bei den subvalvulären Aortenstenose liegt die Einengung unterhalb der eigentlichen Aortenklappe, d.h. im Inneren der linken Herzkammer (= linker Ventrikel), in seinem Ausflußtrakt. • Es gibt 2 Formen dieser angeborenen Herzerkrankung (Abb. 14):
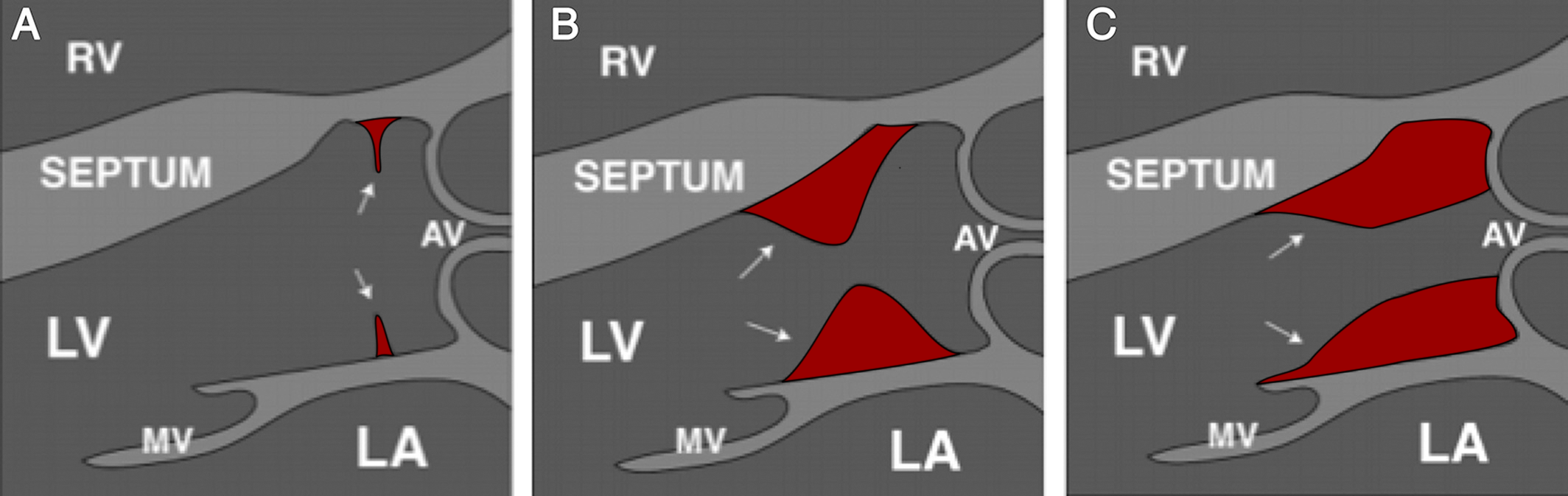 |
| Abb. 14 |
| A: Membranöse Stenose; B: Umschriebener Wulst; C: Langstreckige tunnelförmige Verengung RV = rechter Ventrikel, LV = linker Ventrikel, LA = linker Vorhof; MV = Mitralklappe, AV = Aortenklappe |
- Es hat sich während der Entwicklung des Herzens im Mutterleib eine dünne Membran aus Bindegewebe gebildet („A“ in Abb. 14), die direkt unterhalb der Aortenklappe liebt und den Blutausfluß aus der linken Herzkammer schon vor der Aortenklappe behindert. Dies ist die häufigste Form der subvalvulären Aortenstenose.
- Bei der 2. Form bildet sich während der embryonalen Herzentwicklung entweder ein umschriebener Wulst aus Bindegewebe und Herzmuskel, der den sog Ausflußtrakt des linken Ventrikels umschrieben einengt („B“ in Abb. 14) oder der Muskelwulst entsteht über einen längeren Abschnitt des Ausflußtraktes und bedingt hierdurch dessen langstreckige Einengung („C“ in Abb. 14). Zu dieser 2. Form der subvalvulären Aortenstenose gehört auch die sog. hypertrophe obstruktive Cardiomyopathie (HOCM), die aber in einem anderen eBook über die Herzmuskelkrankheiten besprochen wird.
- Die supravalvuläre Stenose liegt zwischen der Aortenklappe und dem Abgang des Truncus brachiocephalicus, der aus dem Aortenbogen entspringt und für Blutversorgung des rechten Armes zuständig ist und die rechten Halsschlagadern abgibt. Auch diese Verengung kann membranartig sein, umschrieben sein oder eine röhrenförmige Gestalt haben (Abb. 15). Verantwortlich für diese Form der Verengung ist eine Entwicklungsstörung im Truncus arteriosus.
Allen Stenosen gemeinsam ist eine vom Ausmaß der Verengung abhängige Muskelverdickung (= Hypertrophie) des linken Ventrikels.
Auswirkungen
Bei jeder Form der angeborenen Aortenstenose besteht eine Behinderung des Blutausflusses aus der linken Herzkammer. Dies kann 2 Folgen haben:
- Es kann nicht mehr genügend Blut durch die Einengung fließen, sodaß es zu einer verminderten Blut- und damit Sauerstoffversorgung des Körpers mit seinen Geweben und Organen kommt. Dies betrifft jedoch nur die sehr schweren Formen der Aortenstenose. Man bezeichnet dies als „Vorwärtsversagen“ des Herzens.
- Sehr viel häufiger geschieht aber folgendes:
Bei allen Formen der angeborenen Aortenstenose mit der Behinderung des Ausflusses muß der linke Ventrikel einen erhöhten Druck aufbringen, um das Blut durch die Verengung zu pressen. (Bei der hypertrophen Cardiomyopathie (HOCM) ist die Situation etwas komplizierter, sie wird in einem speziellen eBook erklärt.)
Die Notwendigkeit, einen erhöhten Blutdruck aufzubringen hat zur Folge, daß sich der Herzmuskel im linken Ventrikel verdickt (= Hypertrophie).
Diese Hypertrophie ermöglicht es dem Ventrikel einerseits, trotz der Ausflußbehinderung, die ausreichende Menge an Blut in den Kreislauf auszupumpen und (wie die Ärzte sagen:) „kompensiert“ zu arbeiten. Sie erfahren (bei Interesse) mehr hierüber in dem eBook über die Regulation des Kreislaufs.
Zum anderen ändert sich durch die Herzmuskelverdickung aber die Elastizität des Ventrikels und dies hat Auswirkungen auf die Füllung der Kammer:
Normalerweise ist der Herzmuskel mehr oder weniger dünn und damit elastisch. Es ist für das Blut kaum ein Problem, aus dem linken Vorhof durch die Mitralklappe in den linken Ventrikel zu fließen.
Sie können dies etwa mit einem dünnen elastischen OP-Handschuh vergleichen, den Sie unter einen Wasserkran halten, um ihn zu füllen. Das ist kein Problem, denn bei nur dünnem und elastischem Handschuh paßt jede Menge Wasser hinein. Nehmen Sie allerdings keinen OP-, sondern einen gefütterten dickwandigen Winterhandschuh paßt, bei identischer Handschuhgröße, kaum Wasser hinein.
Dieses unterschiedliche Verhalten wird durch die unterschiedlichen Dehnungseigenschaften des Handschuhmaterials verursacht.
Und ebenso ist es beim Ventrikel:
In die normalwandige Kammer paßt sehr viel mehr Blut als in den dickwandigen hypertrophierten Ventrikel. Wenn sich die Kammer aber mit nicht mehr so viel Blut füllen kann staut sich Blut in der linken Vorkammer und ggfs. auch in den Lungen an, weil es halt nicht abfließen kann.
Es kommt also zur Blutstauung und, wenn diese lange genug andauert und schwer genug ist auch zur verminderten Auswurfleistung der Herzkammer. Man spricht in diesen Fällen vom „Rückwärtsversagen“ des Herzens.
Mehr über die verschiedenen Formen der Herzschwäche (= Herzinsuffizienz) erfahren in einem eBook, das sich speziell mit dieser Thematik beschäftigt.
Krankheitserscheinungen
Die körperliche Entwicklung der Kinder ist im allgemeinen ungestört und das Leistungsvermögen erstaunlich groß. Solange das Herz in der Lage ist, alle Gewebe und Organe ausreichend mit Sauerstoff zu versorgen und solange es keine Behinderung des Blutflusses in das Herz hinein, durch das Herz und aus dem Herzen hinaus gibt fehlen in der Regel bedeutsame Beschwerden. Erst dann, wenn der Blutfluß in, durch und aus dem Herzen hinaus behindert und vermindert ist treten Beschwerden wie
- Luftnot,
- Leistungsminderung und
- Herzschmerzen (Angina pectoris) auf.
Bei Kindern mit hochgradiger Stenose fürchtet man den früh eintretende plötzliche Herztod.
Untersuchungsergebnisse
Körperlicher Untersuchungsbefund
(siehe auch eBook über die körperliche Untersuchung)
Patienten mit angeborener Aortenstenose sind körperlich in der Regel unauffällig.
In vielen Fällen haben sie einen eher schlanken und zarten Körperbau.
Bei bedeutsamer Aortenstenose kann man einen verstärkten und hebenden Spitzenstoß palpieren.
Bei schwereren Verengungen fühlt man oft ein Schwirren über dem Herzen und über den Halsschlagadern, was bei den leichteren Formen fehlt.
Geräusch
| Geräusch 1 |
Führendes Symptom aller Aortenstenosen ist ein lautes systolisches Geräusch, dessen punctum maximum (= Stelle, an der man es am lautesten hört), Verlaufsform und Fortleitung in Abhängigkeit von Art und Schwere der Aortenstenose stark variieren kann.
Das Geräusch ist in seinem Beginn vom 1. Herzton abgesetzt, da es erst dann entsteht, wenn Blut durch die Verengung fließt und das Auspumpen des Blutes erst eine kurze Zeit nach dem Beginn des Herzschlages einsetzt. Es handelt sich um ein typisches Austreibungsgeräusch, dessen Maximum in der mittleren bis späten Systole liegt (Geräusch 1).
 |
| Abb. 16 |
| Phonokardiogramm bei angeborener Aortenstenose Oben: Leichte Stenose, spindelförmiges Geräusch mit frühsystolischem Maximum Unten: Hochgradige Stenose, spindelförmiges Geräusch mit spätsystolischem Maximum |
Das Geräuschmaximum liegt in der Regel um so später in der Systole, je hochgradiger die Stenose ist (Abb. 16).
EKG
Siehe auch spezielles eBook.
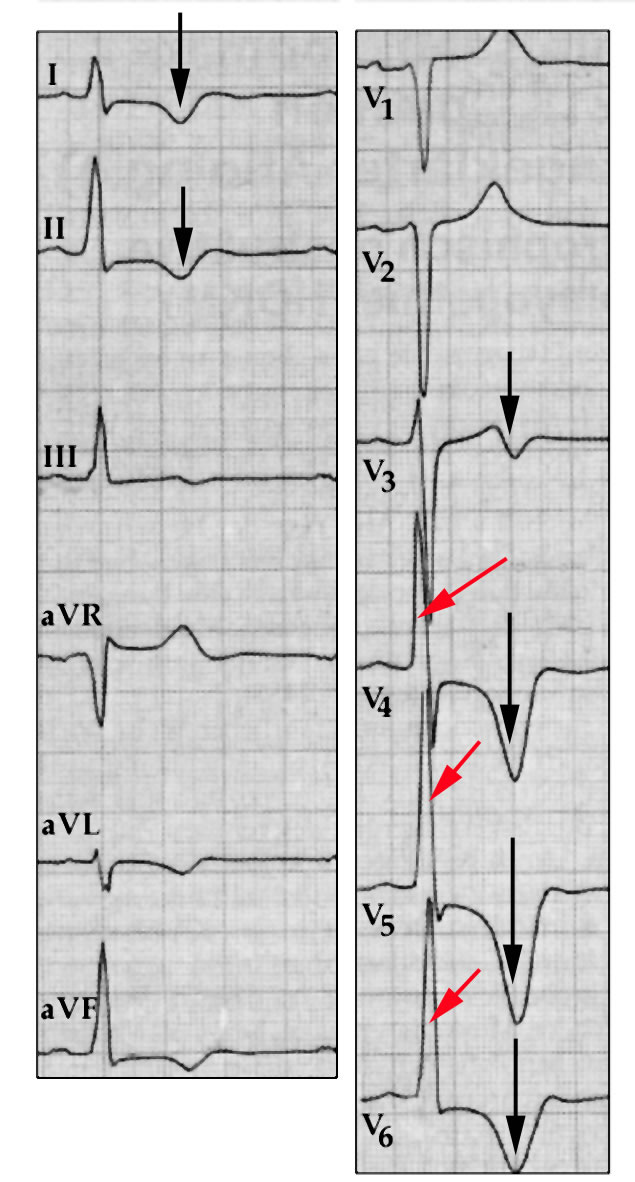 |
| Abb. 17 |
| Schwere angeborene Aortenstenose Die deutliche Verdickung des Herzmuskels der linken Kammer zeigt sich an der Überhöhung der R-Zacken (rote Pfeile), die Schädigung des Muskels an den negativen T-Wellen (schwarze Pfeile). |
Elektrokardiographisch (Abb. 17) sind bei allen stärkeren Formen der Aortenstenose die Zeichen der Verdickung des Muskels der linken Hauptkammer (= linksventrikuläre Hypertrophie) vorhanden.
Zudem sieht man bei den schweren Formen der Aortenstenose auch die Schädigung des Herzmuskels in einer deutlichen Negativierung der sog. T-Welle.
Aus der Schwere der EKG-Veränderungen kann bis zu einem gewissen Grad auf die Schwere der Stenose rückgeschlossen werden. Dies gilt aber vor allem für erwachsene Patienten (siehe eBook über die erworbenen Herzfehler), während dies bei Kleinkindern weniger zuverlässig ist.
Röntgen
Siehe auch spezielles eBook über Röntgenuntersuchungen.
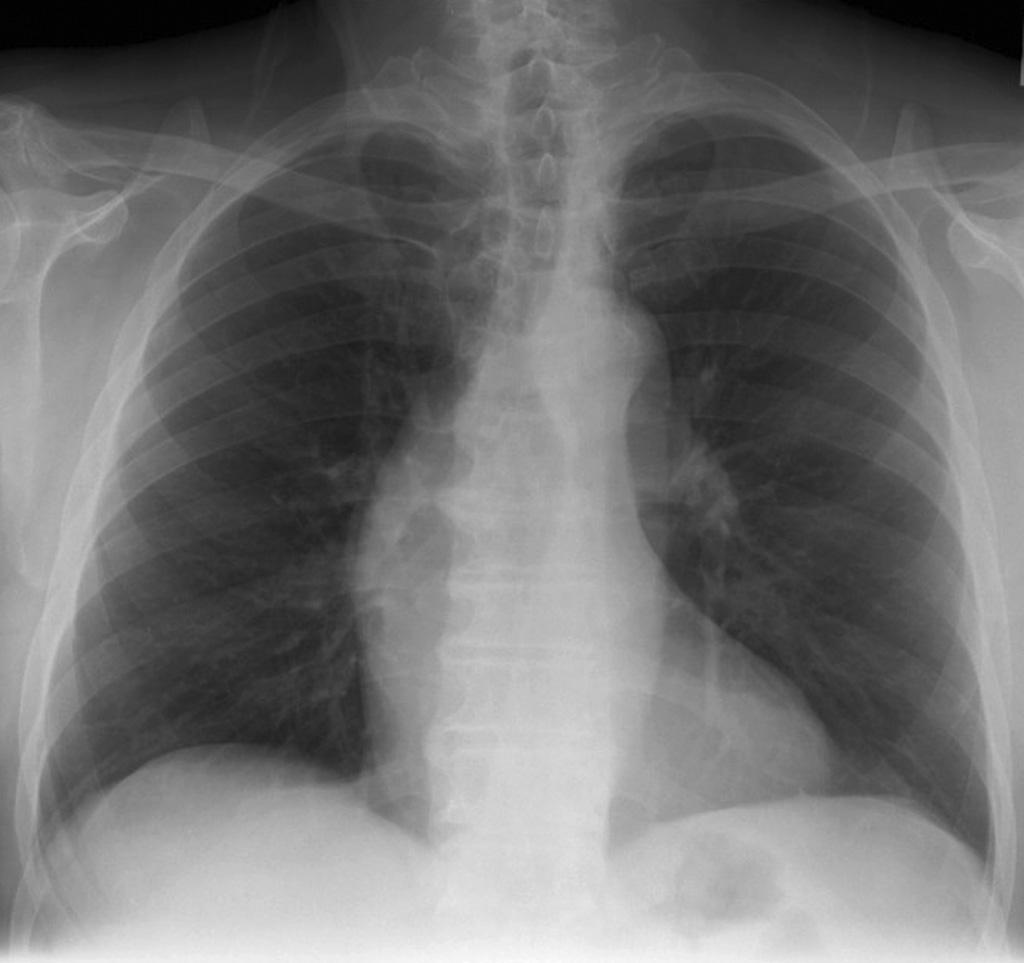 |
 |
|
| Abb. 18 | Abb. 19 | |
| Kompensierte hochgradige angeborene Aortenstenose Nach rechts ausladender Aortenschatten, keine Dilatation des linken Ventrikels (Case courtesy of Assoc. Prof. Frank Gaillard, Radiopaedia.org, rID: 15391) |
Angeborene Aortenstenose mit Herzschwäche |
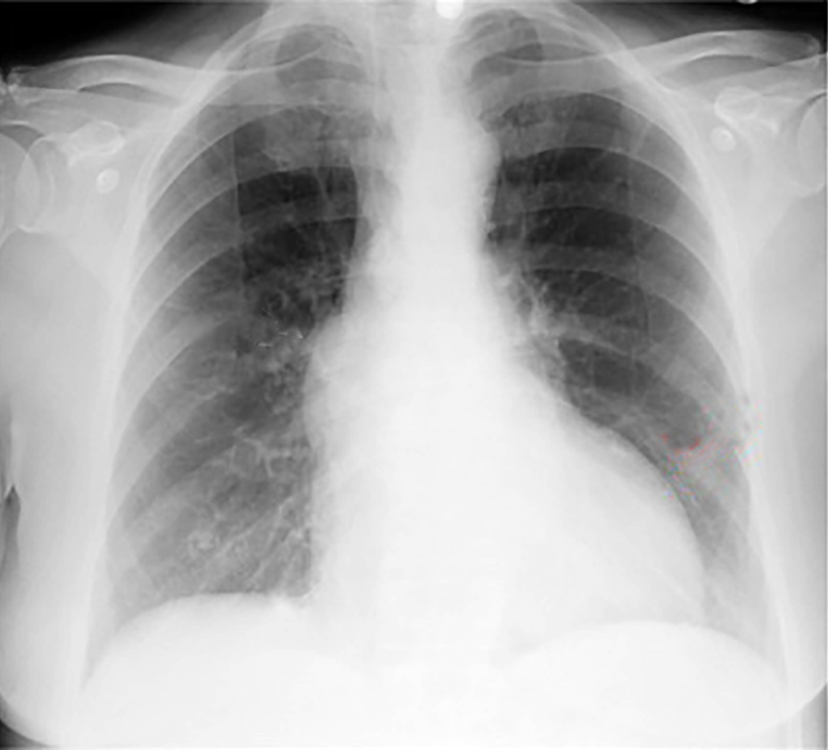
Im Röntgenbild ist das Herz, solange der Herzfehler nicht dekompensiert ist, nur gering vergrößert.
Was die Form des Herzens angeht ist sein linker Rand oft stärker gerundet als normal. Die Aorta ist in ihrem Anfangsteil oft erweitert (Abb. 18).
Diese Erweiterung findet man besonders häufig bei der valvulären Stenose, sie kommt aber bei den anderen Formen des Herzfehlers ebenfalls vor. Bei einer langen röhrenförmigen supravalvulären Stenose ist die Aorta oft nur klein, sodaß sie kaum erkennbar ist. Ist der Herzfehler erst einmal dekompensiert, d.h. ist die Menge des ausgepumpten Blutes vermindert und es kommt zur Blutstauung wird das Herz im Röntgenbild größer (Abb. 19).
Echokardiographie
Siehe auch spezielles eBook über Ultraschalluntersuchungen.
Die Echokardiographie ist eine wichtige Untersuchung für die Diagnose einer angeborenen Aortenstenose, denn man kann mit dem „normalen“ Echo die Art der Aortenstenose feststellen und mit dem DOPPLER-Echo die Schwere des Fehlers messen. Besonders bei Kindern liefert das Echo meistens hervorragende Bilder.
 |
| Abb. 20 |
 |
| Abb. 20 |
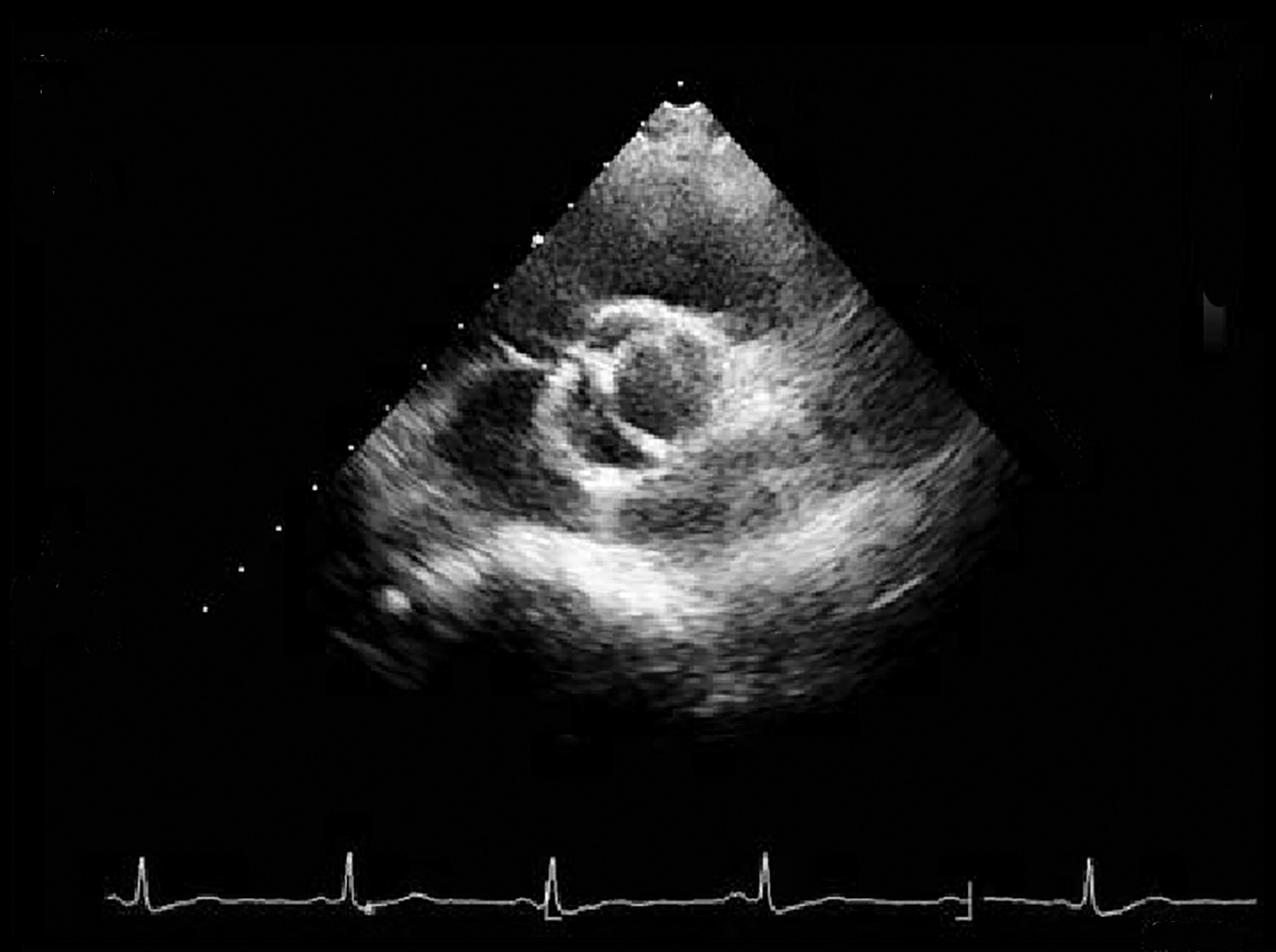
- Bei der valvulären Aortenstenose sieht man, auch bei schon älteren Patienten, die abnormale Klappe mit ihren nur 2 Taschen gut erkennen (Abb. 20).
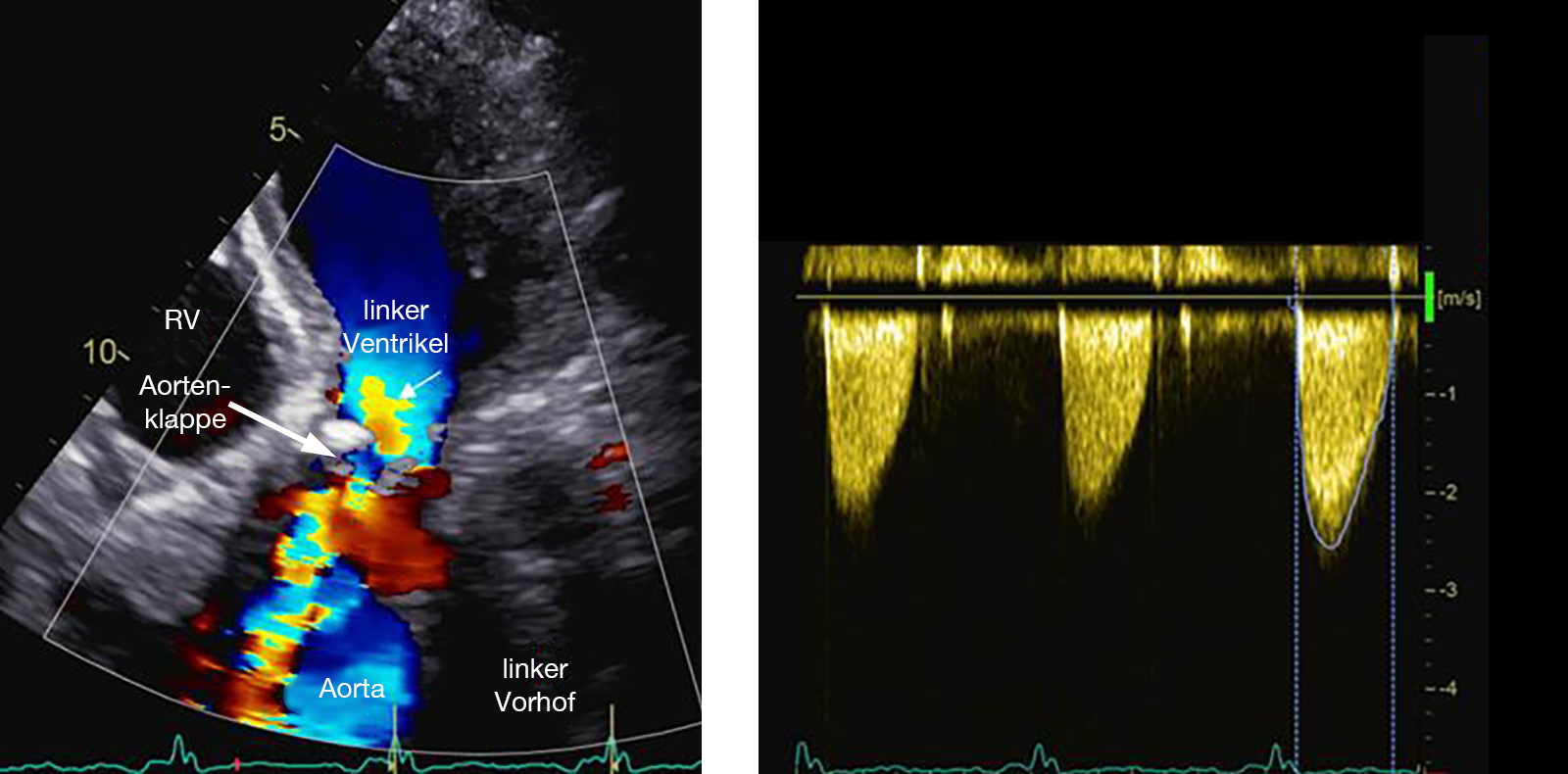
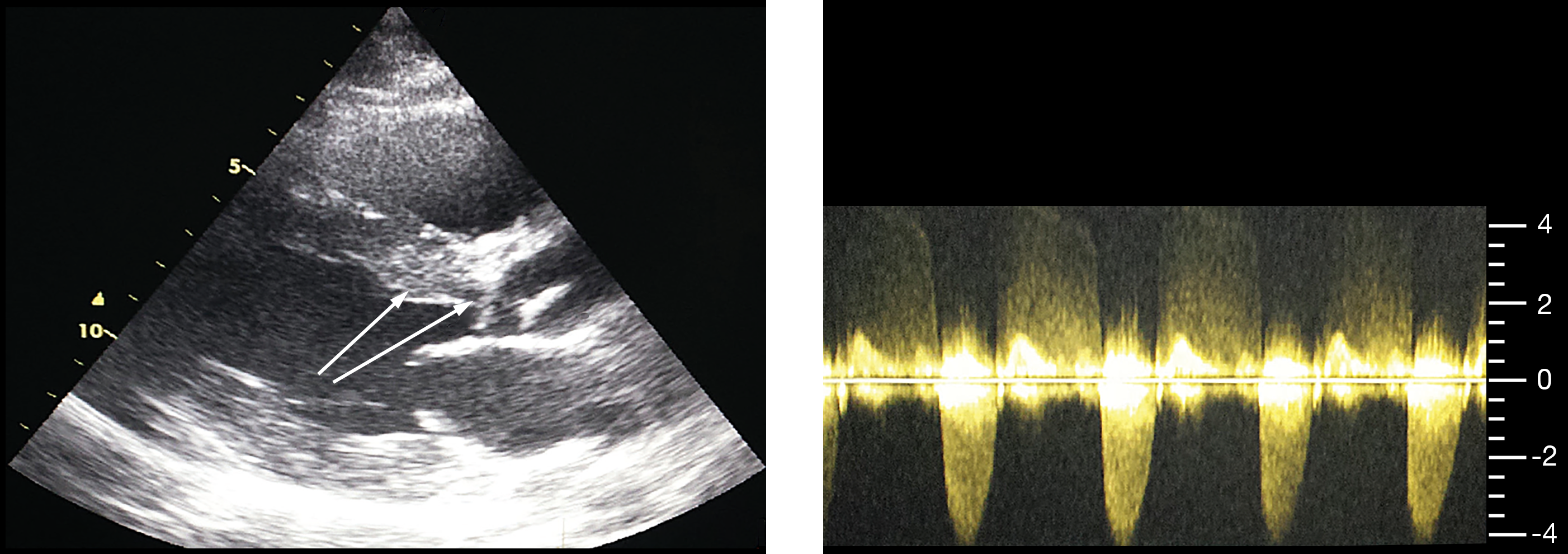
Ist es aufgrund dieser Klappenveränderung zu einer Verengung gekommen kann man im Farb-DOPPLER sehen, wie das Blut an der Klappe verwirbelt wird und mit dem DOPPLER-Echo messen, wie stark das Blut in der Verengung beschleunigt wird, d.h. wie schwerwiegend der Fehler ist (Abb. 21).
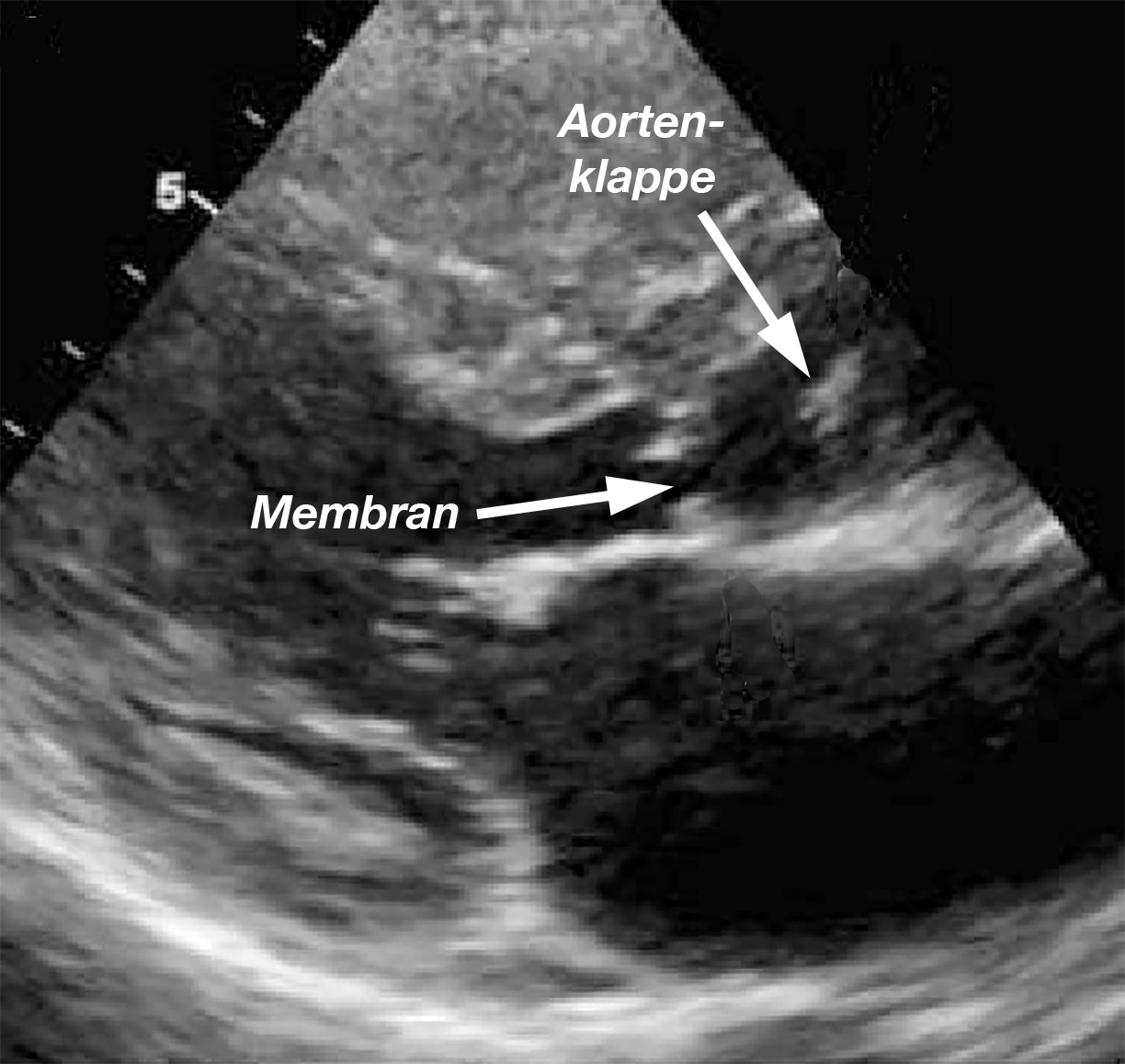
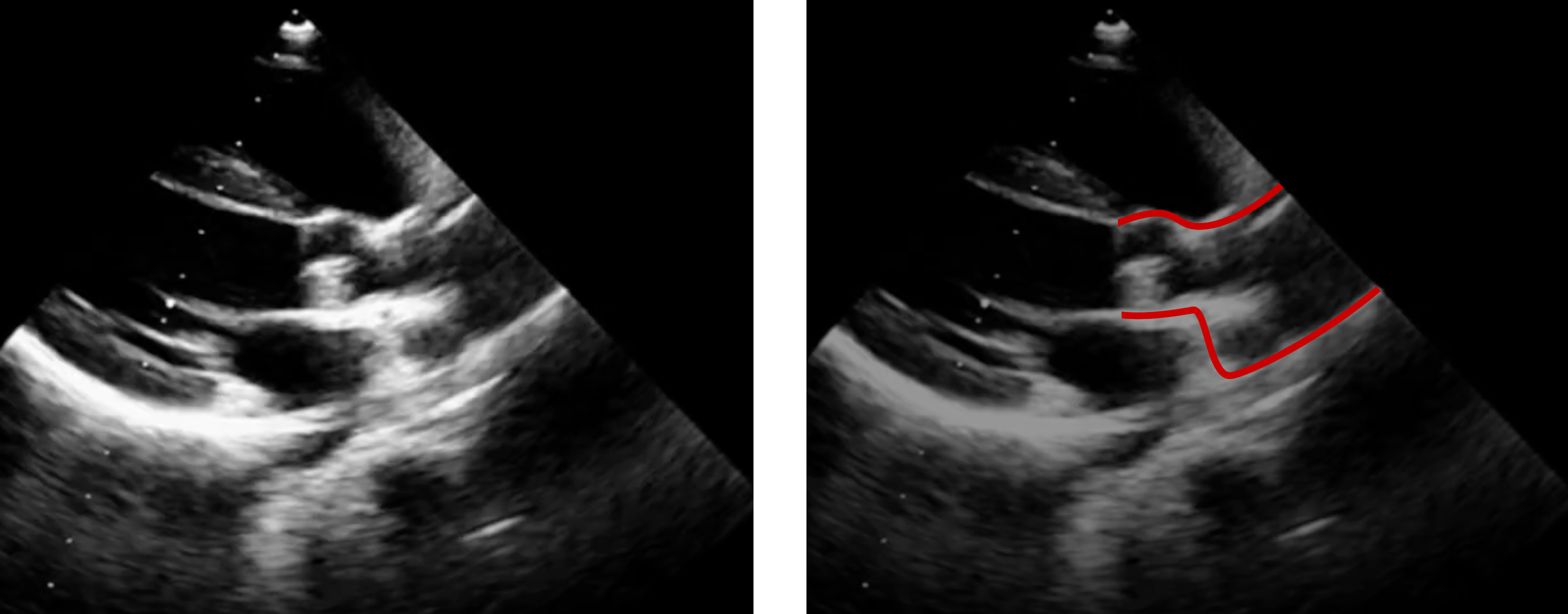
- Bei der subvalvulären Stenose kann man die unterhalb der Aortenklappe gelegene Membran bei Kindern sehr gut erkennen (Abb. 22), bei älteren Kindern und Erwachsenen ist dies kaum möglich. Die sehr viel seltener auftretenden Formen dieses Herzfehlers mit der umschriebenen oder längerstreckigen Verengung aus Herzmuskel- und Bindegewebe ist dagegen im „normalen“ Echo nicht so gut erkennbar (Abb. 23), aber auch hier sieht man im Farb-DOPPLER die Verwirbelung des Blutes in der Engstelle und man kann die Flußgeschwindigkeit im DOPPLER messen.
- Ob man die supravalvuläre Aortenstenose im Echo erkennen kann hängt davon ab, wie weit sie von der Aortenklappe entfernt ist. Umfaßt sie den Ring der Klappe (Abb. 24) oder liegt nur geringfügig von der Klappe selber entfernt kann man sie gut erkennen.
 |
| Abb. 22 |
 |
| Abb. 23 |
| Links: Echo mit längerstreckiger Verengung Rechts: DOPPLER-Echo mit deutliche Beschleunigung des Blutflusses auf ca. 4 m/sec (normal: max. 1 m/sec) |
|
| Abb. 24 |
Je weiter sie von der Klappe entfernt ist desto schlechter kann man sie erkennen, denn die Aorta taucht an diesen Stellen in die Tiefe des Brustkorbes ab und man kann hier nur schwer sehen.
 |
| Abb. 25 Links: Supravalvuläre Aortenstenose, die den Ring der Aortenklappe erfaßt und die sich von hier aus etwas weiter in die Aorta reicht Rechts: Dasselbe Bild wie links, jedoch mit markierter Kontur der Aorta |
Magnetresonanz-Tomographie (MRT)
Siehe auch spezielles eBook über MRT-Untersuchungen.
 |
 |
| Abb. 25 | Abb. 26 |
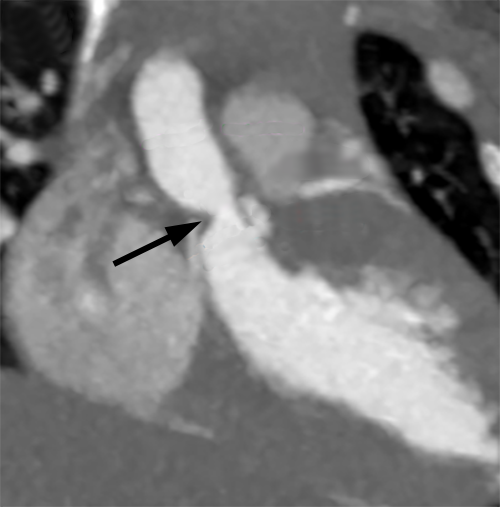
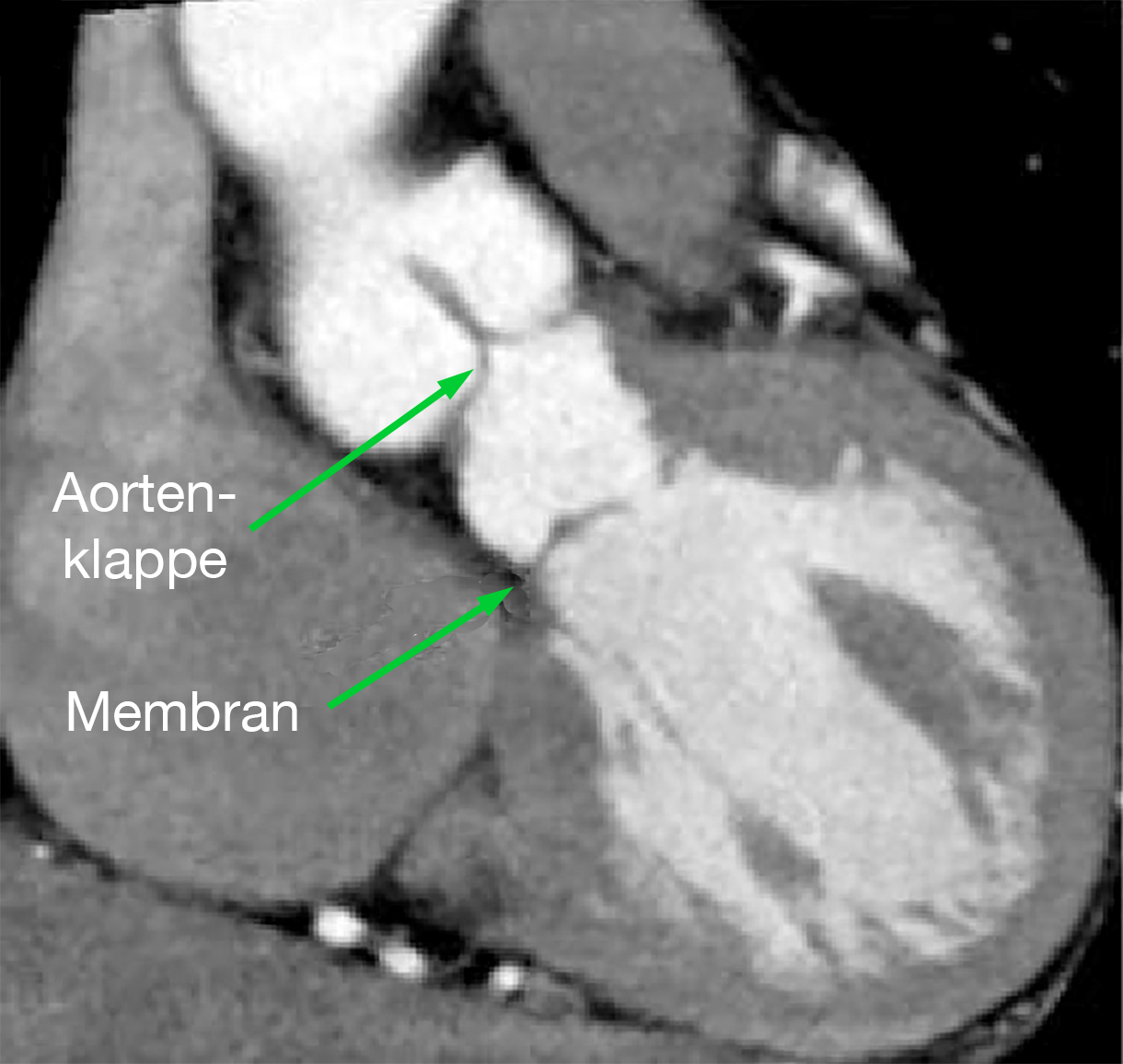
| Supravalvuläre Aortenstenose (Pfeil) im MRT | Membranförmige subvalvuläre Aortenstenose im MRT |
Die MRT-Untersuchung spielt in der Diagnostik eine eigentlich nur untergeordnete Rolle, denn prinzipiell liefert sie dieselben Ergebnisse wie das Echo (Abb. 25 und 26).
Dennoch kann auch ein MRT notwendig werden, nämlich dann,
- wenn das Echo (aus welchen Gründen auch immer) qualitativ nicht gut ist oder
- wenn nach dem Echo noch Fragen offen bleiben, wie z.B. die Längenausdehnung einer supravalvulären Aortenstenose oder der Einbezug der Abgänge der Arm- und Halsschlagadern.
Auch mit der MRT-Untersuchung ist es möglich, ebenso wie beim Echo den Schweregrad des Fehlers zu bestimmen, indem man die Geschwindigkeit des Blutflusses in der Verengung mißt und dann mit dem ebenfalls gemessenen Durchmesser der Aorta die Fläche berechnet, durch die das Blut fließen kann.
Herzkatheter
Siehe auch spezielles eBook über Herzkatheter-Untersuchungen.
Herzkatheteruntersuchungen werden nicht bei jeder angeborenen Aortenstenose durchgeführt, sondern nur
- wenn nach der Echo- und DOPPLER-Untersuchung eine schwerwiegende Verengung vorzuliegen schein (Druckunterschied ca. 50 mm Hg),
- wenn das Kind Luftnot unter Belastung, Brustschmerzen beklagt oder wenn ein Ohnmachtsanfall aufgetreten ist,
- wenn sich das EKG im Laufe der Entwicklung des Kindes in einer speziellen Weise verändert oder
- auch bei beschwerdefreien Menschen, wenn sie intensiven (Hochleistungs-) Sport betreiben möchten.
Im Rahmen von Herzkatheteruntersuchungen sind 2 Dinge wichtig:
- Die Messung des Blutdrucks an verschiedenen Stellen der linken Hauptkammer und
- die Darstellung des Herzens und der Aorta mit einer Kontrastmittelinjektion (= Angiographie).
Druckmessung
Das für die Diagnose einer Aortenstenose entscheidende Kriterium ist der Nachweis einer Druckdifferenz an den Stellen vor und hinter der Verengung.
Wenn man den Katheter beispielsweise aus der Leistenschlagader gegen den Blutstrom durch die Aorta bis in den linken Ventrikel vorschiebt muß man durch die Verengung hindurch. Diese Passage ist bei der supra- und subvalvulären Verengung in vielen Fällen relativ einfach möglich, bei der valvulären Form kann es aber schwierig werden, in allen Fällen hängt die Möglichkeit der Passage vom Ausmaß der Verengung ab.
 |
| Abb. 27 |
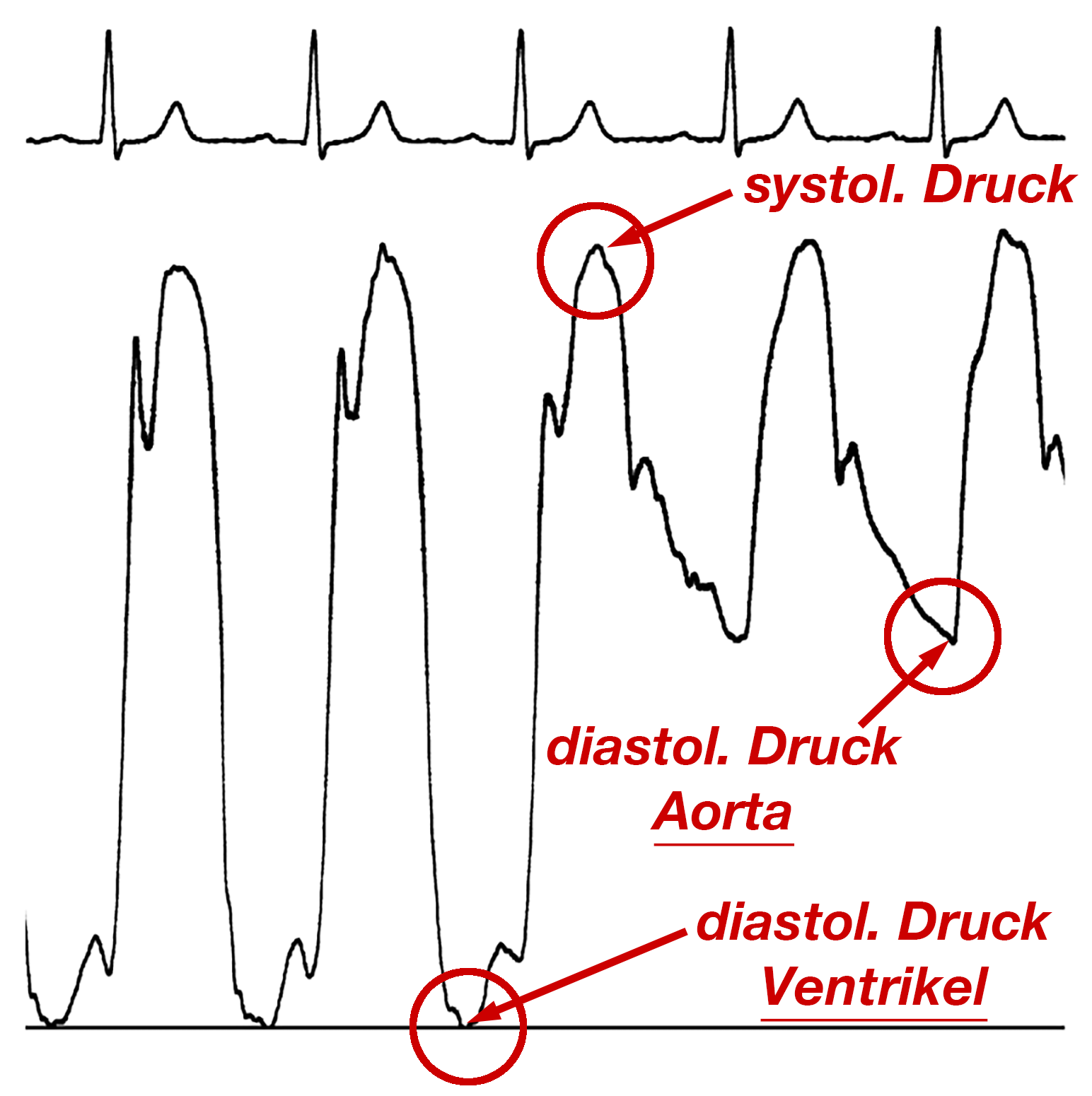
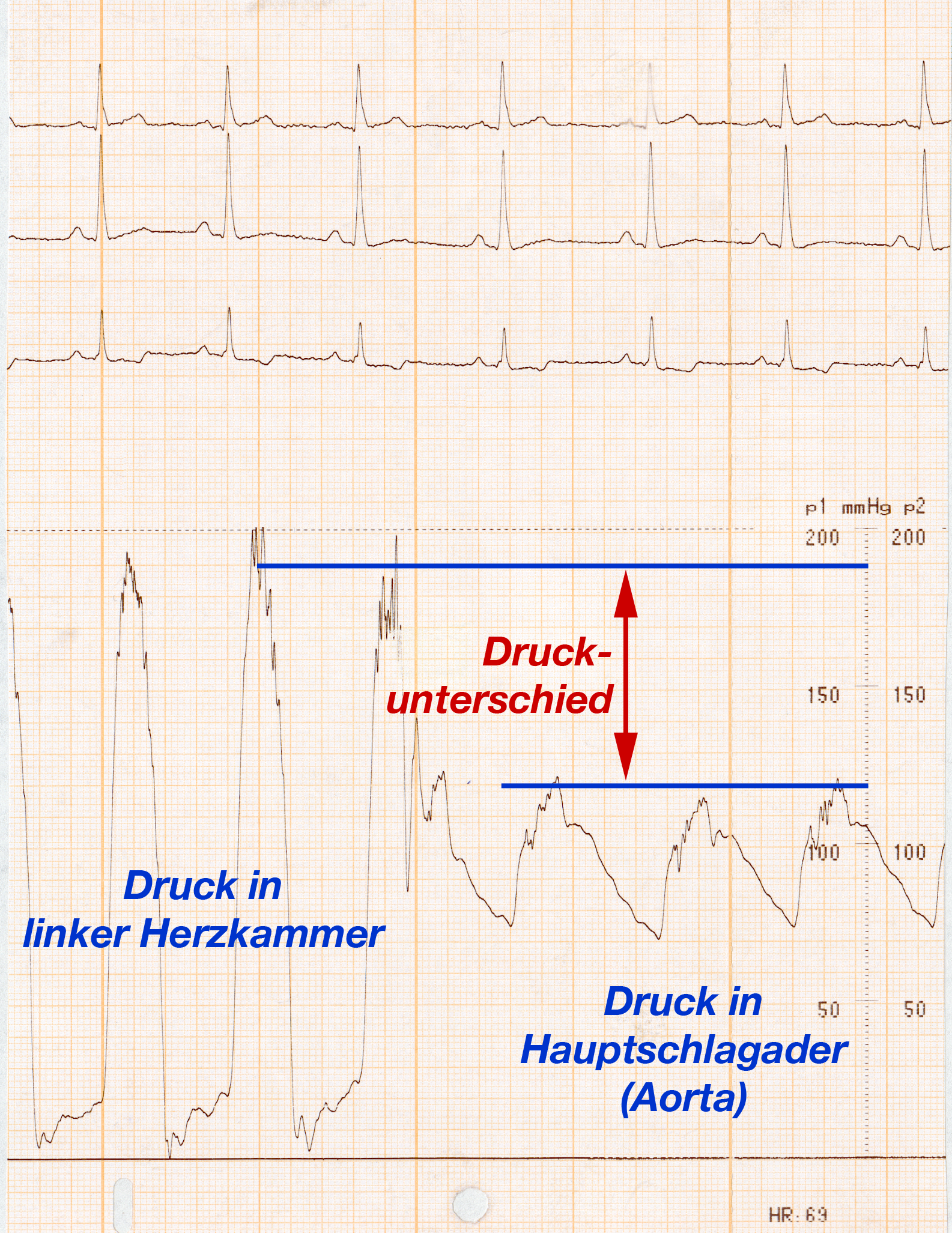
Entscheidend für die Diagnose ist die Druckkurve beim Rückzug aus der Spitze des linken Ventrikels in die Aorta. Dabei gilt, daß der Druck vor der Verengung höher als hinter ihr ist (siehe oben), denn ansonsten könnte ja kein Blut durch die Verengung hindurch fließen.
Aus dem Ort des Drucksprungs kann man die Lage der Stenose (sub-, supra- oder valvulär) erkennen, während das Ausmaß des Drucksprungs Auskunft über den Schweregrad der Verengung gibt. Den Ort, an dem die Katheterspitze liegt, kann man bei Betrachtung der Druckkurvenform erkennen:
Innerhalb des Ventrikels beträgt der niedrigste Punkt der Druckkurve auf 0 bzw. nahezu 0 ab (= diastolischer Druck), jenseits der Aortenklappe ist der diastolische Druck wesentlich höher (Abb. 27).
 |
| Abb. 28 |
| Valvuläre Aortenstenose |
An dieser charakteristischen Formänderung der Druckkurve kann man also erkennen, wann die Katheterspitze auf ihrem Weg vom Ventrikel in die Aorta die Aortenklappe passiert hat. Bezüglich des Ortes des Drucksprungs gibt es die folgende Möglichkeiten:
- Wenn der Druck im linken Ventrikel erst mit der Passage der Aortenklappe absinkt liegt die Verengung in der Klappe selber (= valvuläre Aortenstenose) (Abb. 28).
Sie erkennen den Druck in der linken Herzkammer und in der Aorta, sowie den Übergang vom Ventrikel in die Aorta an dem erhöhten diastolischen Druck in der Aorta. Im Ventrikel ist der systolische Druck deutlich höher als in der Aorta („Druckunterschied“ in Abb. 28).
Das Ausmaß dieses systolischen Druckunterschiedes zeigt Ihnen die Schwere der Verengung an (je größer der Druckunterschied, desto schwerwiegender die Verengung). Und daß es sich um eine Verengung der Klappe handelt sehen Sie daran, daß der Druckunterschied exakt zu demjenigen Zeitpunkt erfolgt, an dem die Katheterspitze aus dem Ventrikel in die Aorta gezogen wird.
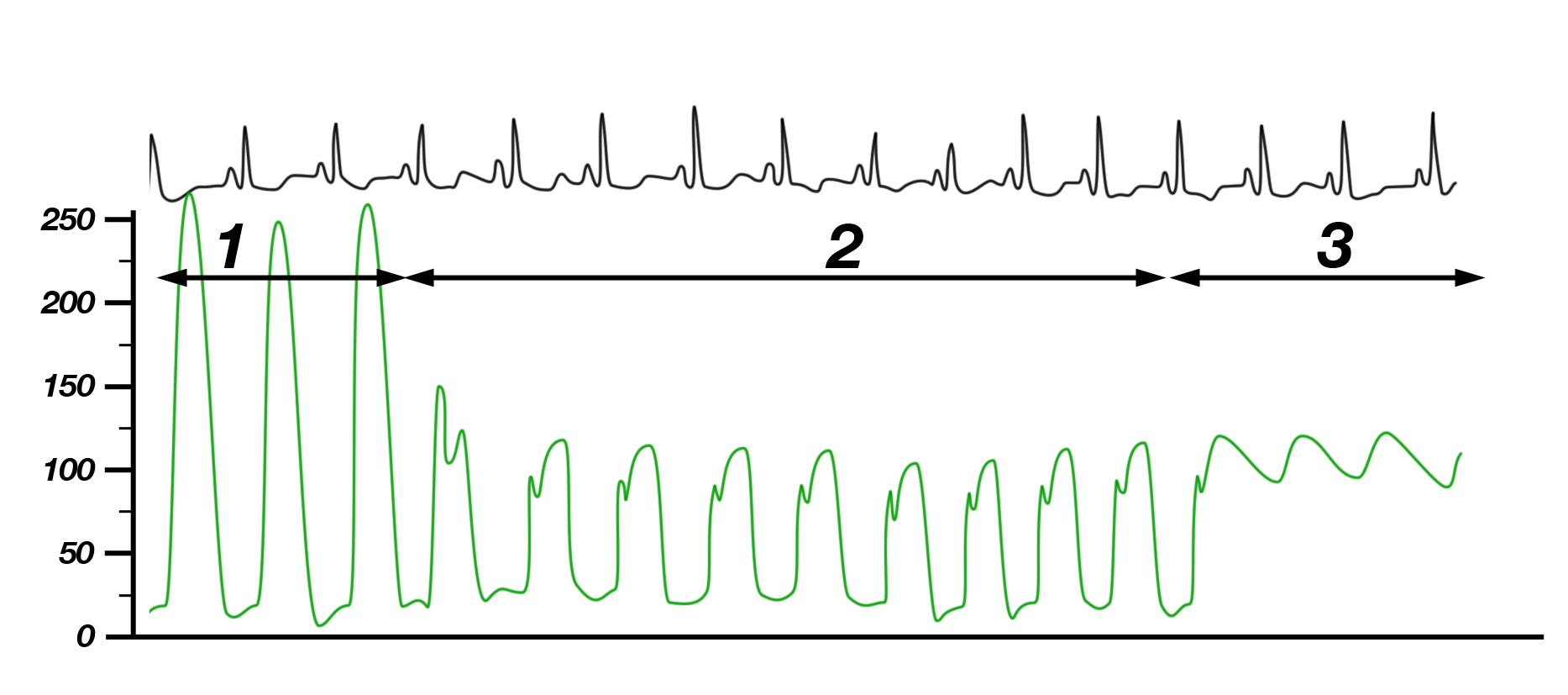
- Demgegenüber zeigt die subvalvuläre Stenose (Abb. 29) eine 2fache Druckänderung:
Sie sehen hier folgendes:
In der Spitze des Ventrikels („1“ in Abb. 29) ist der systolische Druck sehr hoch und der diastolische Druck fast 0.
Zieht man den Katheter etwas zurück fällt der systolische Druck ab („2“), was auf die subvalvuläre Stenose zu beziehen ist, der diastolische Druck bleibt aber mehr oder weniger unverändert niedrig, d.h. der Katheter befindet sich noch innerhalb des Ventrikels.
Bei „3“ schließlich steigt der diastolische Druck schlagartig an, was zeigt, daß sich die Katheterspitze jetzt durch die Aortenklappe in die Aorta bewegt hat. Zwischen „2“ und „3“ ist aber kein Druckunterschied festzustellen, d.h. daß die Aortenklappe nicht verengt ist.
Die Stenose liegt in diesem Fall unterhalb der Aortenklappe, also subvalvulär. Die charakteristische Form der Druckkurve bei der HOCM, einer speziellen subvalvulären Aortenstenose, wird im eBook über die Herzmuskelerkrankungen besprochen.
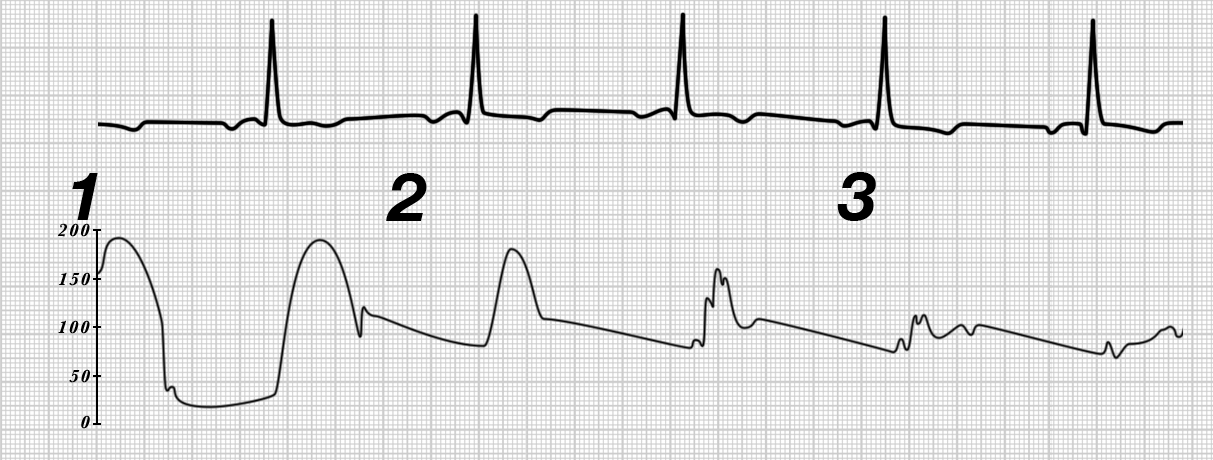
- Und in Abb. 30 sehen Sie schließlich den Katheterrückzug aus dem Ventrikel, durch die Aortenklappe bis etwa in Höhe des Aortenbogens bei einer supravalvulären Aortenstenose.
In „1“ sehen Sie den typischen Ventrikeldruck mit dem niedrigen diastolischen Druck und in „2“ den erhöhten diastolischen Druck, der anzeigt, daß die Katheterspitze nun in der Aorta liegt.
Erst bei „3“ sehen Sie nun einen systolischen Druckunterschied. Dies zeigt an, daß die Verengung, auf der dieser Druckunterschied beruht, hinter der Aortenklappe, also supravalvulär, liegt.
 |
| Abb. 29 |
| Subvalvuläre Aortenstenose |
 |
| Abb. 30 |
| Supravalvuläre Aortenstenose |
Angiographie
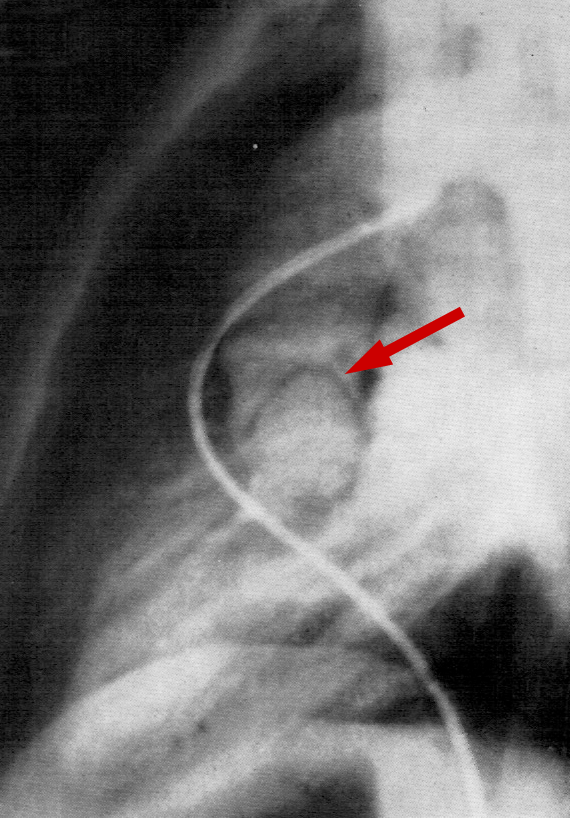 |
| Abb. 31 |
| Angeborene valvuläre Aortenstenose |
Bei den angeborenen Aortenfehlern spielt die Kontrastmitteleinspritzung eigentlich nur eine untergeordnete Rolle, denn Art und Aussehen der Verengung kann man im Ultraschallbild (Echo) und ggfs. dem MRT ebenso gut erkennen.
- Bei der angeborenen valvulären Stenose sieht man eine Aortenklappe, die sich oft domförmig öffnet.
In Abb. 31 ist das Kontrastmittel (KM) in die Lungenschlagader gespritzt worden. Linker Ventrikel, Aorta und Aortenklappe färben sich dann an, nachdem das KM durch die Lunge geflossen ist und das linke Herz erreicht hat.
Abb. 31 zeigt die dünne, d.h. nicht verdickte Aortenklappe, die sich während ihrer Öffnung domförmig (roter Pfeil) entfaltet. Oft ist zudem der Anfangsteil der aufsteigenden Aorta, die Aortenwurzel, deutlich erweitert, was bei den anderen angeborenen Aortenstenosen nur selten auftritt.
- Die supravalvuläre Stenose wird entweder an einer eng umschriebenen Einschnürung des Aortenrohres oberhalb der Klappenebene (Abb. 32, roter Pfeil) oder an einer längeren röhrenförmigen Einengung erkannt, wobei die Aorta nach der Verengung oft erweitert ist (Abb. 32, grüner Pfeil).
Die Koronararterien, die sich aus der vor der Stenose gelegenen Aorta aus mit dem hier herrschenden besonders hohen Druck füllen sind meistens deutlich erweitert.
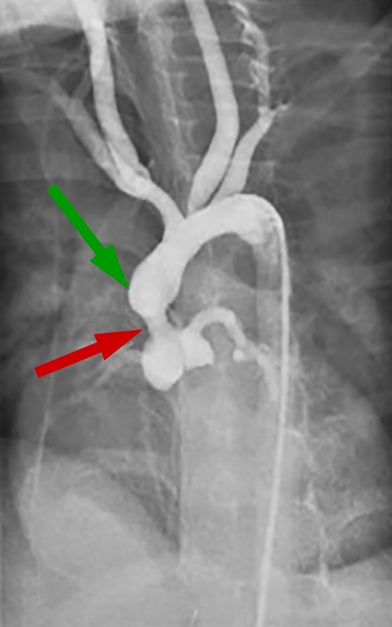 |
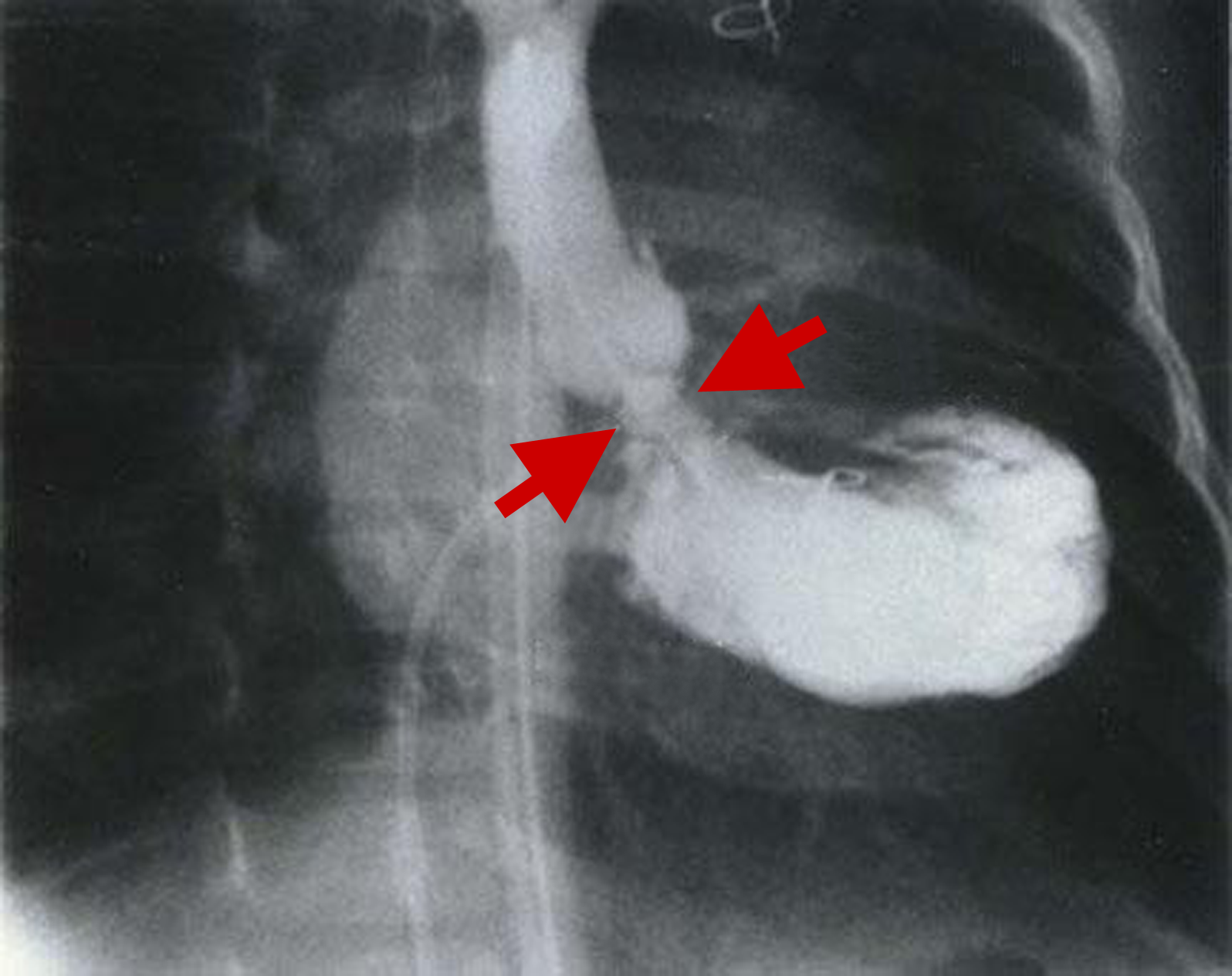 |
| Abb. 32 | Abb. 33 |
| Supravalvuläre Aortenstenose | Subvalvuläre Aortenstenose |
- Für die subvalvuläre Stenose ist im Angiogramm typisch, daß die Verengung nur unterhalb der Aortenklappe besteht und daß man sie am besten während der Pumpphase des linken Ventrikels sieht (Abb. 33, rote Pfeile). Aortenklappe und Aorta sind unauffällig.
Krankheiten mit ähnlichen Erscheinungen
Die angeborenen Verengungen der Aorta treten im frühen Säuglings- und Kindesalter kaum in Erscheinung. Erst im weiteren Verlauf des Kindes treten körperliche Entwicklungsstörungen, Luft, Brustschmerzen oder Herzrhythmusstörungen auf, weil der Körper größer wird, dadurch einen Sauerstoff- und Blutbedarf hat, dieser vermehrte Bedarf aber infolge der Verengung nicht ausreichend bedient werden kann.
Weil die „gesunden“ Teile des Herzens mit zunehmenden Alter wachsen, die bindegewebigen Teile des Herzens, die die Verengung verursachen, jedoch nicht mitwachsen können, nimmt der Herzfehler mit zunehmendem Alter zu. Dennoch wird ein angeborener Aortenfehler nur ganz selten bei Menschen jenseits der Pubertät festgestellt, weil bei den zuvor schon stattfindenden Routineuntersuchungen eines Säuglings und Kindes oft Herzgeräusche festgestellt werden, die dann Anlaß zu weiteren Untersuchungen sind und zur Feststellung des Fehlers führen.
Komplikationen
Komplikationen des Herzfehlers treten dann auf, wenn der Fehler nicht rechtzeitig erkannt und behandelt wurde. In diesen Fällen drohen Herzschwäche mit zunehmender Luftnot und der plötzliche Herztod infolge bösartiger Herzrhythmusstörungen.
Wie bei zahlreichen angeborenen Herzfehlern besteht auch bei den Aortenstenosen die Gefahr einer bakteriellen Endokarditis der Aortenklappe, die zu einer Schädigung der Klappe führen kann.
Notfälle
Bei schwerwiegenden Verengungen kann es, unabhängig davon, welche Aortenstenose vorliegt, zur Dekompensation des Herzens kommen. Dabei ist das Herz nicht mehr in der Lage, den Körper ausreichend mit Blut zu versorgen.
Die Folgen sind Luftnot und Wasseransammlungen in der Lunge (Lungenödem).
Diese Situationen erfordern eine sofortige Behandlung.
Ebenfalls als Notfall ist das plötzliche Auftreten von Ohnmachtszuständen (= Synkope) zu sehen, die in der Regel durch bösartige Herzrhythmusstörungen verursacht werden.
Vorbeugende Maßnahmen
Da es sich um einen angeborenen, genetisch bedingten Herzfehler handelt, ist eine Vorbeugung nicht möglich.
Faktoren, die das Risiko erhöhen, im Laufe des Lebens zu erkranken
Die Erkrankung besteht bereits zum Zeitpunkt der Geburt.
Verhaltensweisen, die die Heilung fördern
Eine Heilung der Erkrankung mit bestimmten Medikamenten oder Verhaltensweisen ist nicht möglich.
Verhaltensweisen, die die Krankheit verschlimmern
Da die Erkrankung bereits Neugeborene betrifft gibt es keine Verhaltensempfehlungen für die Betroffenen selber.
Handelt es sich um eine schwerwiegende Verengung der Aortenklappe, der unterhalb der Klappe im Ventrikel gelegenen Ausflußbahn oder der Aorta jenseits der Aortenklappe ist eine Behandlung mit den unten stehenden Verfahren erforderlich.
Hat die Erkrankung ein nicht so schwerwiegendes Ausmaß muß man mit dem (Kinder-) Kardiologen vor allem anhand des Ultraschallbefundes klären, ob man dem Kind körperliche Schonung (z.B. durch Befreiung vom Schulsport) verordnen muß. Dies ist immer dann notwendig, wenn man befürchten muß, daß stärkere körperliche Anstrengungen (wie das bei Kindern und Jugendlichen nicht zu vermeiden ist) zu einer Überlastung des ohnehin belasteten Herzmuskels mit den entsprechenden Schäden vor allem am Herzmuskel führen.
Bei leichten Formen der Erkrankung ist eine körperliche Schonung aber keinesfalls immer erforderlich.
Therapie
Valvuläre, sub- und supravalvuläre Stenosen werden unterschiedlich behandelt, mit Ballonkathetern oder einer Operation. Eine medikamentöse Behandlung ändert am Ausmaß der Verengung nichts. Sie wird nur eingesetzt, wenn man die Zeit bis zur definitiven Behandlung, der Operation, überbrücken muß. Als Dauerbehandlung bei Menschen mit leicht oder mittelschwerer Erkrankung, die (noch) keiner Operation bedürfen, ist sie ebenfalls ohne größere Bedeutung.
Alle Medikamente, die man zur Behandlung einer (durch andere Erkrankungen bedingten) Herzschwäche einsetzt (z.B. ACE-Hemmer, AT1-Antagonisten oder entwässernde Medikamente) dürfen nur in geringen Dosen und unter fortlaufender, engmaschiger Überwachung im Rahmen einer Krankenhausbehandlung eingesetzt werden, denn sie können schnell zu einer Verschlechterung der Situation führen.
Auch wird die Vorbeugung gegen eine bakterielle Endokarditis (= Endokarditis-Prophylaxe) wird nach heutigem Wissenstand nicht pauschal empfohlen.
Valvuläre Stenose
Die Behandlung der valvulären Aortenstenose erfolgt entweder mit einer Ballonerweiterung der verengten Klappe oder mit einer Operation.
Durchgeführt werden diese Eingriffe bei Kindern oder Heranwachsenden
- bei höhergradigen Verengungen (Druckunterschied im DOPPLER-Echo ca. 50 mm Hg) und
- beim Auftreten von Beschwerden (Brustschmerzen, Ohnmachtsanfall oder Luftnot bei Belastung),
- oder wenn es bei Verlaufsbeobachtungen zu bestimmten EKG-Veränderungen kommt,
- bei Verengungen mit einem Druckunterschied von >60 mm Hg auch bei beschwerdefreien Patienten.
Ist eine Behandlung erforderlich hat man hierfür die folgenden Möglichkeiten:
Ballonerweiterung
 |
| Abb. 34 |
Man benutzt hierzu einen Ballonkatheter (Abb. 34).
Der Katheter mit dem Ballon an seiner Spitze wird über die Leisten- oder (bei Neugeborenen) über die Nabelarterie soweit in Richtung auf das Herz vorgeführt, bis er mitten in der verengten Aortenklappe liegt. Zur genauen Positionierung wird die Position des Ballons während des Eingriffs mit Ultraschall beobachtet.
Der Ballon wird, wenn er seine Position erreicht hat, mit einer Mischung aus Kochsalzlösung und Kontrastmittel mit Druck „aufgeblasen“. Hierdurch werden die verwachsenen Klappentaschen auseinander-„gerissen“ und die Öffnungsfläche der Klappe hierdurch erweitert.
Es ist, bevor man sich zu dieser Behandlung entschließt, unbedingt erforderlich, 3 Dinge abzuklären:
- Es sollte sich um eine Aortenklappe handeln, die mindestens 2 Taschen besitzt. Bei den angeborenen Aortenstenosen liegt aber oft nur 1 einzige Tasche vor. In diesen Fällen liefert die Ballonerweiterung keine guten Ergebnisse, denn diese einzige Tasche wird durch die Ballonerweiterung oft zerrissen und es entsteht eine bedeutsame Undichtigkeit der Klappe.
- Der Rand der fehlgebildeten Aortenklappe sollte möglichst kreisrund und nicht, wie es bei diesen Fehlern aber oft vorkommt, exzentrisch sein. Auch hier droht nach der Ballonerweiterung eine bedeutsame Klappenundichtigkeit.
- Es muß geklärt werden, worin genau das Problem an der Aortenklappe besteht: Ist „nur“ die Klappe mißgebildet oder ist der Ring, in dem die Klappe angebracht ist, verengt. Eine Ballonerweiterung ist lediglich dann möglich, wenn die Klappe selber das Problem darstellt. Ist der Klappenring verengt ist eine Operation erforderlich.
Bei ca. 20 - 25% aller Patienten, die mittels Ballonerweiterung behandelt werden muß man innerhalb 1 Jahres, in ca. 30% innerhalb von 5 Jahren, 50% innerhalb von 10 Jahren und ca. 70% innerhalb von 20 Jahren mit dem Auftreten einer erneuten Verengung rechnen, die sich im weiteren Verlauf des Lebens entwickelt. In diesen Fällen ist eine Operation (s.u.) oft unumgänglich (25 - 30% innerhalb von 10 Jahren).
Operation
Alle nachfolgend erwähnten Operationen finden unter Vollnarkose und unter Benutzung der Herz-Lungen-Maschine statt (bzgl. der Herz-Lungen-Maschine siehe eBook über eine Herzklappen-Operation).
Hinsichtlich der Operation bestehen mehrere Möglichkeiten:
- Klappen-„Reparatur“: Immer dann, wenn die Aortenklappe selber das Problem ist, z.B. weil 2 fehlgebildete Klappentaschen miteinander verwachsen sind, kann der Chirurg versuchen, die Klappe zu reparieren.
Dazu wird nach Eröffnung des Brustkorbes die Aorta knapp oberhalb der Herzklappe freigelegt. Durch einen kleinen Einschnitt in die Aortenwand kann der Chirurg nun „von oben“ auf die Klappe sehen und sie behandeln.
Er kann, je nach Zustand der Klappe, evtl. miteinander verwachsene Klappentaschen mit dem Skalpell voneinander trennen und er kann dicke Bindegewebs-Auflagerungen und -Knötchen entfernen.
Ein in dieser Technik erfahrener Chirurg erzielt mit dieser Operationstechnik oft gute Erfolge, aber dennoch muß man bei ca. 50% aller Patienten innerhalb von 10 Jahren mit einer erneuten Aortenklappenoperation und bei ca. 30% mit einem Klappenersatz (s.u.) rechnen.
Gelingt es nicht, die Klappe zu reparieren, etwa weil die Klappentaschen infolge ihrer Fehlentwicklung zu stark mißgebildet sind, weil der Klappenring zu klein ist, weil sich nach einer zuvor durchgeführten Ballonerweiterung erneut eine Verengung gebildet hat oder wenn es nach einer vorherigen Ballonerweiterung zu einer bedeutsamen Undichtigkeit der Klappe gekommen ist bleibt nur die Möglichkeit eines Klappenersatzes.
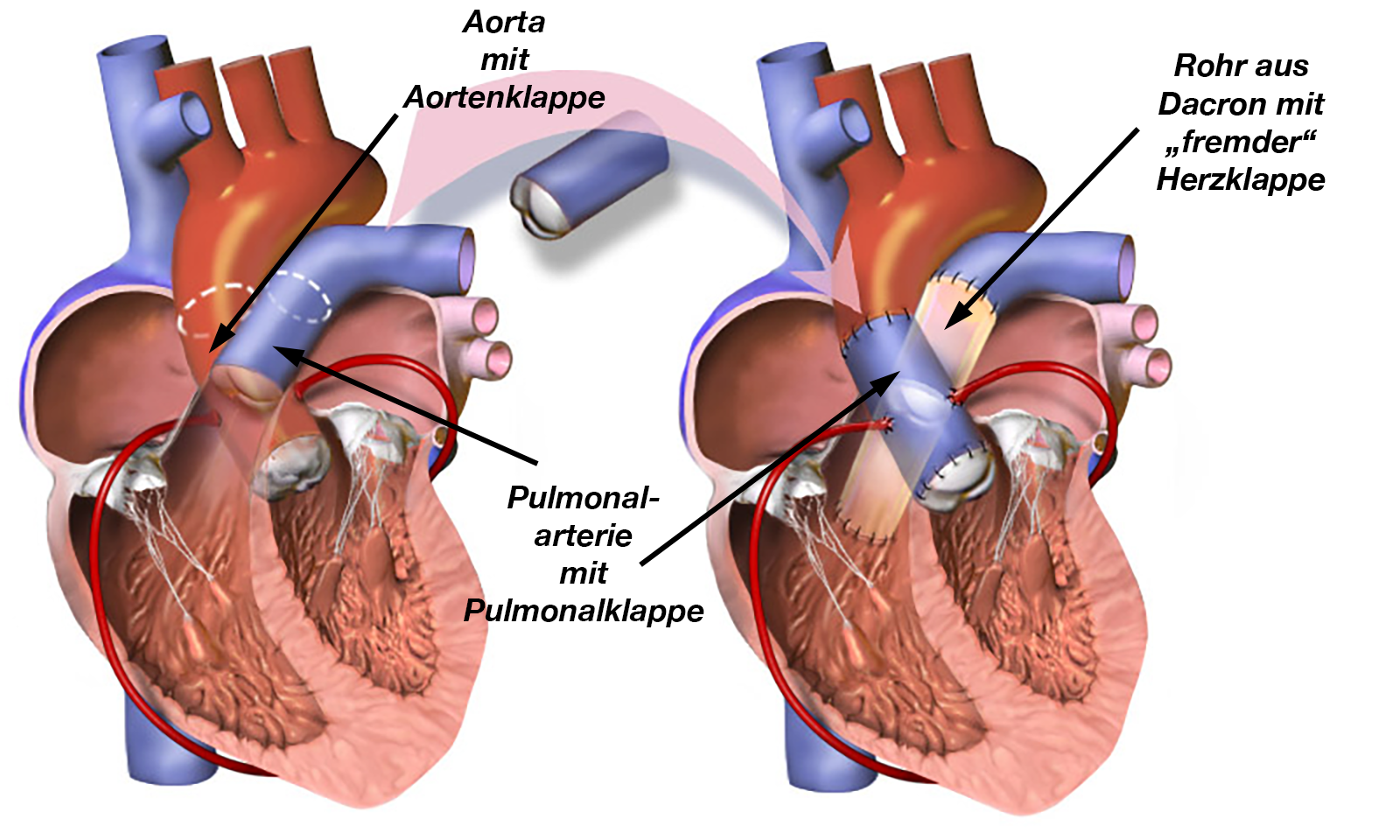
- Klappenersatz: Auch hier gibt es mehrere Möglichkeiten: Nach der Entfernung der mißgebildeten Klappe implantiert man eine mechanische Klappenprothese, eine biologische Klappe oder man führt den ROSS-Eingriff durch.
- Der Einsatz einer mechanischen Klappenprothese ist nur bei größeren Kindern oder Erwachsenen möglich, weil der Durchmesser des Aortenklappenrings hier groß genug ist, um eine Klappenprothese mit guten Durchflußmöglichkeiten zu implantieren.
Die mechanische Klappenprothese hat aber den Nachteil, daß sie nicht mit dem größer werdenden Kind mitwachsen kann. Zudem ist es erforderlich, daß Kind lebenslang mit blutverdünnenden Medikamenten (Marcumar) zu behandeln.
Die Operation hat mit einer Sterblichkeit von ca. 5% eine geringes Risiko. Man kann damit rechnen, daß innerhalb von 10 Jahren bei 20 - 25% eine erneute Operation erfolgen muß (ca. 1%/Jahr), vor allem bei Patienten, die zum Zeitpunkt der Operation sehr jung waren und die daher eine nur schmale Aorta haben.
- Eine solche Blutverdünnung ist bei Verwendung einer biologischen Klappenprothese nicht notwendig. Allerdings kann auch eine solche Prothese nicht mit dem größer werdenden Kind mitwachsen. Zudem haben biologische Klappen eine nur begrenzte Lebensdauer (ca. 10 - 15 Jahre), sodaß sie im weiteren Leben des Kindes erneut chirurgisch ausgetauscht werden müssen: Ca. 40% innerhalb von 12 Jahren.
- Die 3. Möglichkeit zur Operation wird ROSS-Eingriff genannt.
Hierbei wird die mißgebildete Aortenklappe entfernt und gegen die Lungenklappe (Pulmonalklappe) desselben Patienten ausgetauscht. Die Pulmonalklappe selber wird durch die Spenderklappe eines anderen Menschen, die in ein Rohr aus Dacron eingesetzt wird, ersetzt (Abb. 35).
Eine der Voraussetzungen ist logischerweise, daß der rechte Teil des Herzens gesund ist und/oder daß kein weiterer Herzfehler bzw. eine Erkrankung vorliegt, die diesen Teil des Herzens ebenfalls betrifft.
Die Vorteile dieser Operation sind die fehlende Notwendigkeit zur blutverdünnenden Behandlung und die Möglichkeit der „neuen“ Klappen, mit dem Kind mit zu wachsen. Aus diesen Gründen wird diese Operationstechnik heute sehr häufig angewandt.
Das Operationsrisiko einer ROSS-Operation ist in Kliniken, die große Erfahrung mit dieser Technik haben, gering. Wenn Säugling und kleine Kinder mit der ROSS-Technik operiert werden, geht man davon aus, daß etwa 40% im Verlauf der folgenden 10 Jahre erneut operiert werden müssen, werden ältere Kinder operiert sind dies nur etwa 10%. Daher versucht man, eine ROSS-Operation nach Möglichkeit in die spätere Kindheit bzw. die Pubertät zu verschieben.
Die ROSS-Technik hat aber auch gewisse Risiken, die darin bestehen, daß sich im weiteren Verlauf des Lebens eine Undichtigkeit der „neuen“ Aortenklappe entwickelt und daß es dazu kommen kann, daß sich die Wand der „neuen“ Aortenklappe krankhaft erweitert (= Dilatation).
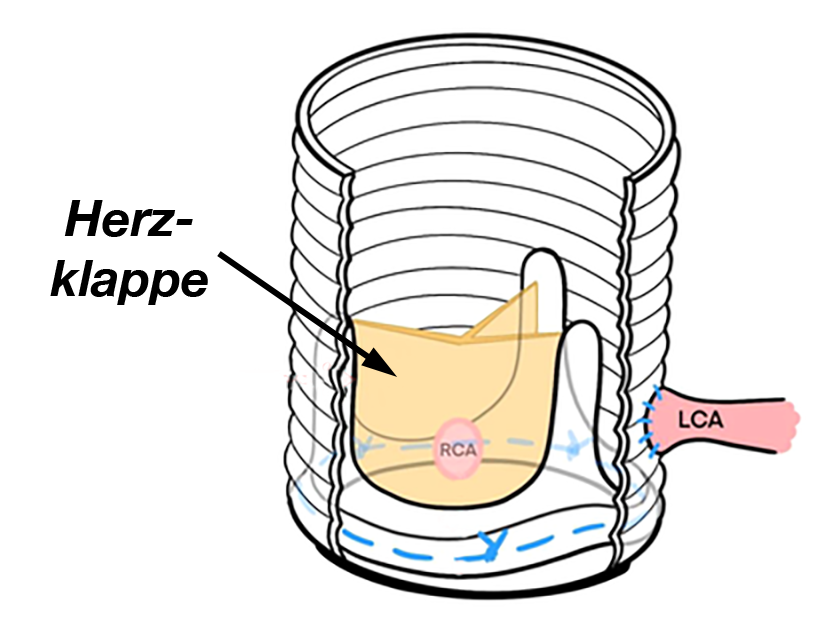
Abb. 36 LCA = linke Koronararterie
RCA = rechte KoronararterieAus diesen Gründen ummantelt man das Stück Wand der Pulmonalarterie, in der die Klappe aufgehängt ist und das zusammen mit der Klappe in die Aorta eingepflanzt wird mit einer Manschette aus Dacron (Gewebe aus feinen Kunststofffasern) (klappentragendes Conduit, Abb. 36).
Dabei müssen die beiden Herzkranzarterien (linke und rechte Koronararterie) neu in die Dacron-Manschette eingepflanzt werden.
Problematisch wird der Eingriff dann, wenn die Aorta an derjenigen Stelle, an der die neue Klappe eingesetzt werden soll zu weit oder zu eng ist. Ist der Klappenring zu weit benutzt man ein Rohr aus Dacron mit der darin eingesetzten neuen Aortenklappe (= Conduit, siehe Abb. 36), entfernt die „alte“ Klappe mit der sich daran anschließenden Aorta und verbindet das Herz und die an dieser Stelle wieder normal weiten Aorta mit dem Conduit.
 |
| Abb. 35 |
| Aus Wikipedia |
Wann sollte man eine Ballonerweiterung und wann eine Operation durchführen?
Diese Frage stellt sich natürlich nur dann, wenn eine echte Alternative besteht.
Aus den oben schon genannten Gründe kann nicht jede valvuläre Aortenstenose mittels Ballonerweiterung behandelt werden und es gibt in diesen Fällen keine Alternative zur Operation. Dies betrifft vor allem Patienten, bei denen sich im Verlauf der Entwicklung eine Undichtigkeit der Aortenklappe entwickelt oder solche, bei denen die Art der Klappenmißbildung keine Ballonerweiterung zuläßt.
Ganz allgemein gilt, daß der Anteil von operierten Patienten, die innerhalb von 5 Jahren erneut an der Aorta operiert werden müssen, mit ca. 35% deutlich geringer ist als diejenigen, die nach einer Ballonerweiterung mittels erneuter Ballonerweiterung oder Operation behandelt werden müssen (ca. 35%).
Ebenfalls ganz allgemein gilt, daß die Frage Ballonerweiterung oder Operation vom Alter des Patienten abhängt, an dem die Behandlungsnotwendigkeit festgestellt wird, von der Schwere des Fehlers und von den anatomischen Gegebenheiten.
Bei Säuglingen und jungen Kindern wird man, sollte dies notwendig sein, zunächst zur Ballonerweiterung tendieren, um Kind und Herz größer werden zu lassen und um eine Operation bei einem möglichst großen Herzen durchzuführen, weil die Ergebnisse einer Operation (s.o.) bei größeren Kindern besser sind.
Supra- und subvalvuläre Stenose
Bei den anderen beiden Formen der angeborenen Aortenstenose ist eine Ballonerweiterung nicht erfolgreich, sodaß hier primär operiert werden muß.
- Bei der subvalvulären Stenose ist es erforderlich, daß das Gewebe, das den Ausfluß des Blutes aus dem Herzen behindert, chirurgisch entfernt wird. dieser Eingriff erfordert seitens des Chirurgen große Erfahrung, denn er darf nicht zu wenig Material aus der Wand der linken Hauptkammer entfernen, aber auch nicht zuviel (siehe Operation der HOCM im eBook über die Herzmuskelerkrankungen).
- Bei der supravalvulären Aortenstenose gibt es 2 Möglichkeiten:
- Bei begrenzten und umschriebenen Verengungen der Aorta kann der Chirurg nach einem Einschnitt in die Aortenwand und dem Einpflanzen eines Stücks Dacron die Aorta erweitern.
- Bei längerstreckigen Verengungen, die mehr oder weniger den gesamten Umfang der Aorta erfaßt haben (= tubuläre Verengung) wird die Aorta in ihrem gesamten fehlgebildeten Anteil entfernt und durch ein Rohr aus Dacron ausgetauscht (siehe auch später unter der Therapie der Aortenisthmusstenose).
Wann muß der Hausarzt aufgesucht werden?
Hier ist zu unterscheiden zwischen Kontrolluntersuchungen und solchen, die beim Auftreten bestimmter Beschwerden erforderlich sind.
- Beim Auftreten von Luftnot unter Belastung, von Herzstolpern oder gar bei plötzlichen Schwindel- oder Ohnmachtsanfällen sollte der Hausarzt unverzüglich aufgesucht werden. Wenn der Herzfehler bereits bekannt sein sollte ist es allerdings sinnvoll, direkt und ohne Umweg den Kinder- oder Erwachsenenkardiologen oder evtl. sogar die kinderkardiologische Ambulanz eines spezialisierten Krankenhauses aufzusuchen, denn hier muß vor allem anhand eines Ultraschallbefundes über das weitere Vorgehen entschieden werden.
- Bei Patienten mit leichter bis mittelschwerer Aortenstenose sollten mindestens 1mal jährlich kinderkardiologische Kontrolluntersuchungen mit EKG und Echokardiographie durchgeführt werden.
Diesen Patienten kann erlaubt werden, sich sportlich leicht zu betätigen, solange keine oder nur eine leichte Verdickung der Ventrikelwände im Echo, keine EKG-Veränderungen feststellbar sind, wenn ein Belastungs-EKG ein normales Blutdruckverhalten unter Belastung zeigt und der Patient keine Beschwerden hat.
- Patienten mit schwerer, aber noch nicht mittels Ballonerweiterung oder Operation behandlungsbedürftigen Erkrankung sollten etwa alle 3 - 6 Monate kontrolliert werden, sie sollten keinen kompetitiven Sport (= Wettkampfsport) betreiben.
- Wenn die betroffenen Kinder älter werden sollten sie auch weiterhin regelmäßig vom Kinderkardiologen beobachtet werden, denn mit zunehmendem Wachstum kann es zu Undichtigkeiten der Aortenklappe kommen. Hier muß dann anhand evtl. Beschwerden, von EKG- und Echo-Befunden darüber entschieden, ob weiter abgewartet oder operiert werden muß.
- Auch Patienten, die einer Ballonerweiterung unterzogen worden sind, bedürfen regelmäßiger Kontrollen mit EKG, Echokardiographie und ggfs. MRT-Untersuchung, denn es kann bei diesen Patienten zu einer erneuten Verengung der Klappe kommen.
In solchen Fällen kann, solange keine bedeutsame Undichtigkeit der Aortenklappe vorliegt, ggfs. eine erneute Ballonerweiterung durchgeführt werden. Sollte sich allerdings erneut eine hochgradige Verengung entwickeln wird in der Regel eine Operation notwendig.
- In einigen Fällen können Patienten mit angeborener Aortenstenose auch das Erwachsenenalter erreichen. Bei ihnen ist es wichtig, daß sie für den Rest ihres Lebens von einem Kardiologen überwacht, kontrolliert und betreut werden. Dies ist zunächst Aufgaben des Kinderkardiologen, der aber dabei hilft, daß der Patienten später von einem erfahrenen (!) Erwachsenen-Kardiologen betreut wird.
Aortenisthmusstenose
Beschreibung der Erkrankung
Unter einer Aortenisthmusstenose im engeren Sinne versteht man eine Einengung des Aortenrohres zwischen dem Ursprung der linken Hals- und Armarterie (A. subclavia sinistra) und der Mündung des Ductus arteriosus BOTALLI (der Ductus BOTALLI ist eine Gefäßverbindung zwischen der Lungenarterie und der Aorta, die für den Kreislauf des Kindes im Mutterleib von entscheidender Bedeutung ist (siehe gesondertes Kapitel später in diesem eBook).
Die Entstehung der Aortenisthmusstenose steht im Zusammenhang mit der embryonalen Entwicklung der Aorta und mit dem Ductus arteriosus.
Embryonale Entwicklung der Aorta
|
| Abb. 36 |
| Schema des Kreislaufes des ungeborenen Kindes Beachten Sie den Ductus arteriosus (BOTALLI) |
Die Entwicklung der Aorta hatte ich schon im allgemeinen Teil über die Embryologie des Herzens beschrieben. Sie wissen, daß sich der Ductus arteriosus während der Entwicklung der Aorta aus dem 6. linken Kiemenbogenarterie entwickelt. Er verbindet die Aorta mit der Lungenschlagader, was während des Lebens des ungeborenen Kindes im Mutterleib aus den eingangs genannten Gründen lebensnotwendig ist (Abb. 37).
Mit der Geburt jedoch ist diese Verbindung nicht mehr nötig und verursacht sogar Probleme, wie Sie in einem der folgenden Abschnitte über den offenen Ductus arteriosus lernen werden.
Damit sich der Ductus mehr oder weniger pünktlich mit der Geburt verschießt ist seine Wand speziell gebaut: In ihrer Wand werden nämlich die Substanzen Prostaglandin (PGE2) und Prostazyklin ......
Ende der Leseprobe von Band 1
Lesen Sie in den eBooks (padBook, phoneBook, Kindle) mehr über:
- die Aortenisthmusstenose und
- den offenen Ductus arteriosus BOTALLI und
- die EBSTEIN-Anomalie.
Auch bei diesen angeborenen Herzfehlern wird u.a. erklärt, wie sich die jeweilige Herzstruktur embryonal entwickelt hat, welche Auswirkungen der Herzfehler hat, welche Beschwerden er verursacht, mit welchen Methoden er untersucht wird und welche Ergebnisse diese Untersuchungsmethoden haben, welche Krankheiten mit ähnliuchen Beschwerden es gibt, welche Komplikationen und Notfälle auftreten können, wie der Herzfehler behandelt wird und wann man damit den Hausarzt aufsuchen solte.
Sie können Band 1 dieser eBook-Reihe bekommen, wenn Sie hier klicken:
- padBook (für iPad und epub3-fähige eBook-Reader)
- phoneBook (für smartPhones)
- Paperwhite (für Kindle Paperwhite)
Wenn Sie hier klicken gelangen Sie zu Band 2.