Es handelt sich um eine seltene Erkrankung, die die kleinen und mittelgroßen Arterien und Vene betrifft. Sie wird durch anti-Neutrophile zytoplasmatische Antikörper (ANCA) verursacht.
Bei diesen Antikörpern handelt es sich um Eiweiße, die aufgrund einer Autoimmunerkrankung gebildet und gegen körpereigene, in diesem Fall vaskuläre Strukturen gerichtet sind. Die Folge sind nekrotisierende granulomatöse Entzündungen der kleinen und mittleren Blutgefäßen wie Arterien, Arteriolen, Kapillaren , Venolen und Venen.
Die Erkrankung wurde 1951 von den beiden Ärzten Jacob Churg und Lotte Strauss entdeckt, weshalb sie zunächst CHURG-STRAUSS-Syndrom genannt wurde. In späteren Jahren wurde sie zusammen mit der WEGENER´schen Granulomatose und der Polyangiitis den granulomatösen Vaskulitiden zugeordnet und erhielt den Namen „allergische Granulomatose mit Polyangiitis“.
Allen diesen Erkrankungen ist die Entzündung von Blutgefäßen gemeinsam und bei allen 3 Erkrankungen kann der Nachweis der ANCA positiv sein.
Der autoimmunologische Charakter der Erkrankung zeigt sich am Vorhandensein einer Hypergammaglobulinämie, erhöhtem IgE-Spiegel, Rheumafaktoren und dem Nachweis der ANCA im Blut.
Wie bei Autoimmunerkrankungen üblich ist die Ursache der Erkrankung nicht bekannt.
Das CHURG-STRAUSS-Syndrom kann aber eine seltene Komplikation bei Patienten mit Asthma bronchiale sein, die Steroid-bedürftig sind oder bei solchen Patienten auftreten, die mit entzündungshemmenden Antagonisten gegen Leukotriene-Rezeptoren (z.B. Montelukast) behandelt werden und bei denen bei erfolgreicher Therapie die orale Steroiddosis reduziert wird.
Betroffen sind auch Patienten, deren Asthma bronchiale von oralen auf inhalative Steroide umgestellt wurde.
Man erklärt sich das Auftreten der Erkrankung in beiden Fällen durch das Absetzen bzw. die Dosisverminderung des systemisch wirksamen Steroids, wodurch es zur Demaskierung des schon „schlummernden“ CHURG-STRAUSS-Syndroms kommt.
Epidemiologie
In Deutschland schätzt man eine Inzidenz von etwa 1 - 2 Fälle / 1.000.000 Einwohner / Jahr. Dabei sind Männer etwas häufiger betroffen als Frauen.
Bei seiner Manifestation sind CHURG-STRAUSS-Patienten meistens 40 - 50 Jahre alt, Männer und Frauen erkranken im Mittel gleichermaßen häufig.
Morbidität und Mortalität sind hauptsächlich durch kardiale Komplikationen (Myokarditis und Myokardinfarkt) bedingt.
Prognose
Die 5-Jahres-Überlebensrate ohne Therapie beträgt etwa 25%.
Die Sterblichkeit innerhalb von 5 Jahren nach Diagnosestellung beträgt etwa 25%, bedingt durch direkte oder indirekte Auswirkungen bzw. Komplikationen der Vaskulitis an den verschiedenen Organen. Das größte Risiko zu versterben tragen dabei Patienten mit schwerer myokardialer und gastrointestinaler Vaskulitis.
Unter Therapie überlegen 90% aller Erkrankten 1 Jahr und 62% 5 Jahre.
Differentialdiagnose
Angesichts der Möglichkeit, daß die Erkrankung aufgrund ihres Charakters viele Organe betreffen und in unterschiedlicher Form in Mitleidenschaft ziehen kann ist die Differentialdiagnose logischerweise sehr umfangreich. Nachfolgend finden Sie eine alphabetische Aufstellung, die aber keinen Anspruch auf Vollständigkeit erhebt:
Akute mesenteriale Ischämie
Akute Nephritis
Aspergillose
Asthma bronchiale
bakterielle Pneumonie
Eosinophilie
Eosinophilie Gastroenteritis
Eosinophilie Leukämie
Eosinophilie Alveolitis
GOODPASTURE-Syndrom
Hypereosinophiles Syndrom
Infektiöse Endokarditis
Kälteagglutininkrankheit
Leukozytoklastische Vaskulitis
Medikamenten-induzierte Eosinophilie
Mikroskopische Polyangiitis
Polyarteritis nodosa
rasch progrediente Glomerulonephritis
Segmentale Glomerulonephritis
WEGENER´sche Granulomatose
Verlauf der Erkrankung
Die Erkrankung verläuft typischerweise in 3 Phasen:
Sie beginnt mit allergischen Symptomen wie Heuschnupfen und allergischem Asthma bronchiale.
In der Folge kommt es zu einem deutlichen Anstieg der Eosinophilen im Blut (>700/µl) mit eosinophilen Infiltraten der Lungen und im Magen-Darm-Trakt.
Am Ende schließlich stehen granulomatöse Entzündungen der kleinen und mittleren Blutgefäße („granulomatöse Vaskulitis“). Üblicherweise tritt diese 3. Phase der Erkrankung etwa 3 Jahre nach dem Auftreten des Asthmas auf, kann sich aber durchaus auch erst mehrere Dekaden nachfolgend entwickeln.
Weil es sich um eine Erkrankung der kleineren und mittelgroßen venösen und arteriellen Blutgefäße handelt ist der Befall prinzipiell jedes Organs möglich.
Die Erkrankung tritt in 2 verschiedenen Phänotypen auf, wobei die Ursache für diese beiden Formen nicht bekannt ist. Es handelt sich dabei um:
den primär vaskulitischen Typ mit Lungen- und Nierenbeteiligung. Er ähnelt der mikroskopischen Polyangiitis.
den primär eosinophilen Typ (beispielsweise mit kardialer Beteiligung), der vor allem zur eosinophilen Infiltration mehrerer Organe führt.
Es ist der Befall einzelner Organe, der zu verschiedenen „Begleit-Erkrankungen“ mit der entsprechenden Symptomatik führt.
Diagnostik
Angesichts der Vielzahl der möglicherweise betroffenen Organe sind verschiedene Fachbereiche an Diagnostik und Therapie des CHURG-STRAUSS-Syndroms beteiligt. Dies bezieht sich sowohl auf den ersten Arztkontakt eines Patienten mit seiner primären Symptomatik als auch auf die erweiterte Diagnostik und die Verlaufsuntersuchungen. Weil die kardiopulmonalen Auswirkungen der Erkrankung die größte Bedeutung haben spielen Pneumologe und Kardiologe eine zentrale Rolle. Daneben sind aber auch Hals-Nasen-Ohren-Ärzte, Rheumatologe, Gastroenterologe und Urologe involviert.
Das American College of Rheumatology (ACR) benennt 6 Kriterien für die Diagnose der Erkrankung. Sind ≥4 Kriterien erfüllt besteht eine Sensitivität von 85% und eine Spezifität von 99% für das Vorliegen der Krankheit. Diese Kriterien sind:
Asthma bronchiale, Heuschnupfen
eine Eosinophilie von mehr als 10% im peripheren Blutbild
chronische eosinophile Sinusitis mit Nasenpolypen
(u.U. flüchtige) Lungeninfiltrate
Histologischer Nachweis einer Vaskulitis mit extravaskulärer Ansammlung von Eosinophilen und
Mono-, Polyneuritis oder Polyneuropathie
Anamnese
Der Befall der verschiedenen Organe hat organspezifische „Komplikationen“ zur Folge, die sich in der entsprechenden Symptomatik äußern.
Die häufigsten Beschwerden, die in der Anamnese angegeben werden und bei denen es sich um frühe Hinweise auf die Erkrankung handelt sind allergischen Symptomen wie
Heuschnupfen und
allergisches Asthma bronchiale.
In dieser Phase tritt regelmäßig eine „B-Symptomatik“ auf mit
starker Abgeschlagenheit
Nachtschweiß
Fieber
ungewollter Gewichtsabnahme
Muskel- und Gelenkschmerzen, die in ihrer Lokalisation manchmal oft täglich wechseln.
Die im weiteren Verlauf auftretenden organspezifischen Komplikationen verursachen die für die jeweilige Komplikation typischen Symptome. Die sind
am Herzen: Myokarditis, Kardiomyopathie, (konstriktive) Perikarditis, Herzinfarkt infolge des Verschlusses von Koronararterien, sowie Tachyarrhythmien (ca. 50%) und evtl. lebensbedrohliche Arrhythmien
an den oberen Atemwege und der Lunge: Paranasale Sinusitis, allergische Rhinitis, Asthma bronchiale, eosinophile Alveolitis, Husten und Hämoptysen
am Magen-Darm-Trakt: Schleimhautulcera, Diarrhoe, Erbrechen, Koliken, Magen-Darm-Blutungen, Symptome einer gastrointestinalen Vaskulitis
an der Haut: Petechien, Purpura, Knötchen in der Haut, ulceröse Nekrosen
an den Nieren: Glomerulonephritis, Niereninsuffizienz mit Protein- und Mikrohämaturie, renale Hypertonie
am peripheren Nervensystem: Polyneuropathie, Mononeuritis multiplex. In schweren Fällen Lähmungserscheinungen von Armen und Beinen und
am zentralen Nervensystem: Apoplex (sehr selten)
Körperliche Untersuchung
Ebenso wie in der Anamnese beschrieben findet man bei der körperlichen Untersuchung neben dem unspezifischen Fieber die Befunde der organspezifischen Komplikationen:
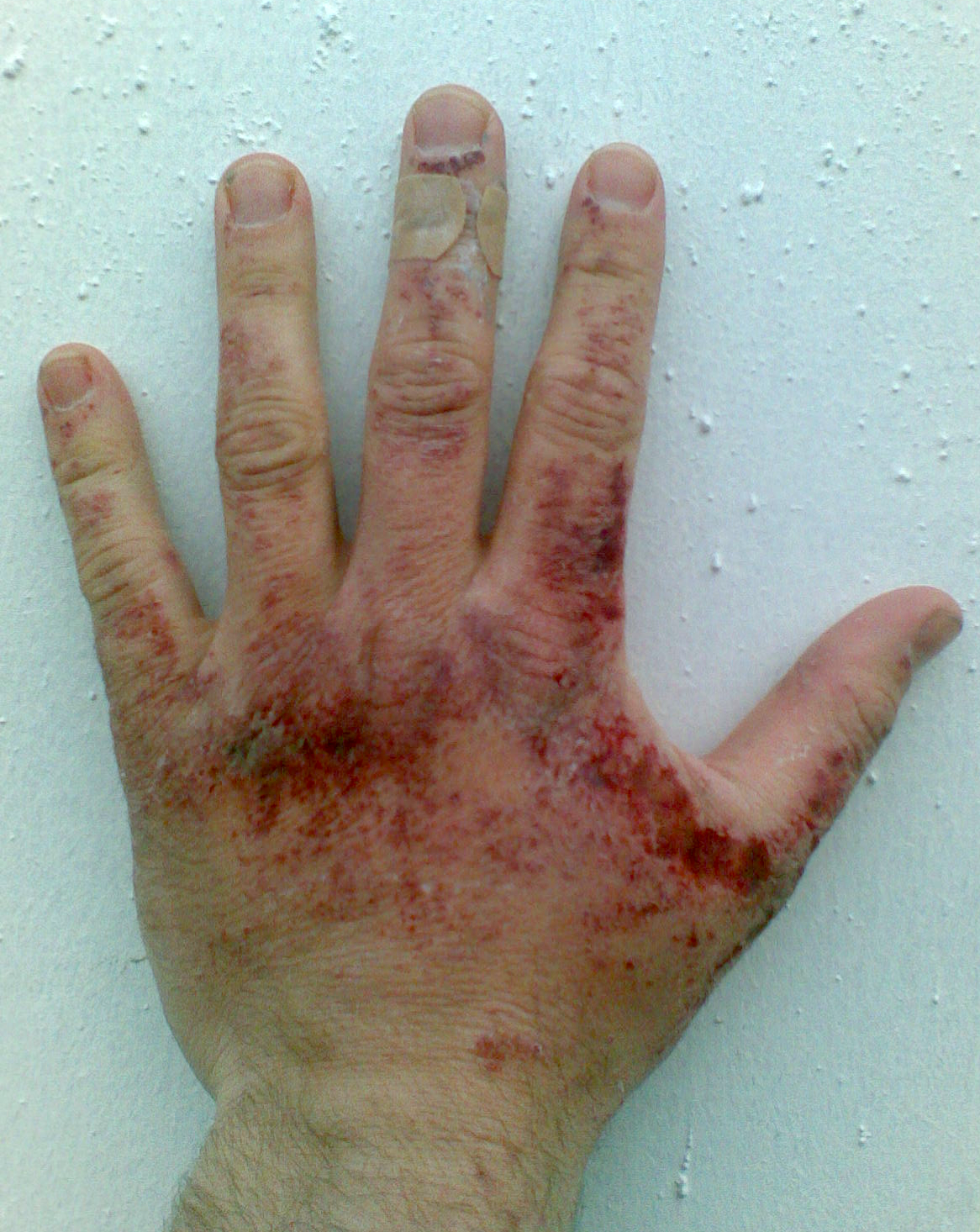
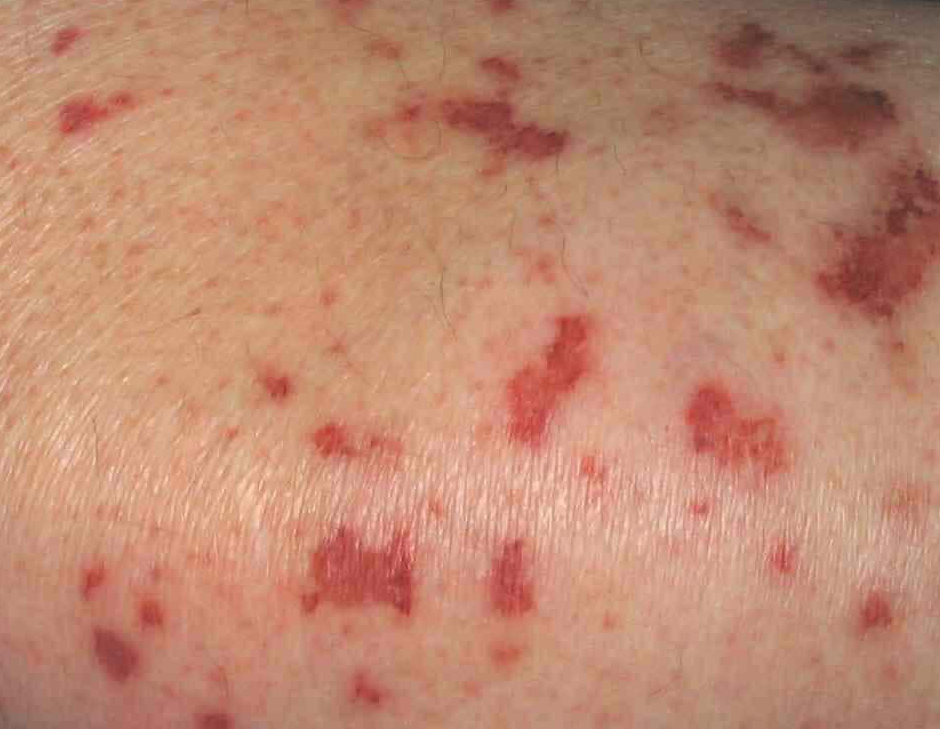
An der Haut: Purpura, Urticaria, Livedo reticularis (Abb. 1), Hautnekrosen oder subcutane Knötchen (Abb. 2)
Abb. 1: Levido reticularis
Abb. 2: Haut-Knötchen
An den oberen Atemwege:
Allergische Rhinitis
Paranasale Sinusitis
Nasen-Polypen
An den unteren Atemwege: Sie auskultieren und sehen die Befunde von
Asthma bronchiale
Pneumonie
evtl. Hämoptysen (infolge alveolärer Blutung bei Alveolarkapillitis)
am Herzen:
Myokarditis und Zeichen einer Herzinsuffizienz
Myokardinfarkt
an den Nieren:
arterielle Hypertonie
Urämie und Zeichen der fortgeschrittenen Niereninsuffizienz
am Gastrointestinaltrakt:
Blutung
Gastroenteritis
Appendicitis
Pancreatitis
akutes Abdomen infolge Darmischämie oder -perforation
am Nervensystem:
periphere Neuropathie
(selten) Apoplex
Apparative Untersuchungen
Laboruntersuchungen
Blut:
Eosinophilie
Anämie
Erhöhung der Blutsenkung (BSG) und des C-reaktiven Proteins (CRP)
Erhöhtes Kreatinin
Antineutrophile Antikörper (ANCA in 70%)
Erhöhtes IgE
Erhöhung von CK und Troponin
Hypergammaglobulinämie
Positive Rheumafaktoren bereits bei niedrigem Titer
Urin:
Proteinurie, mikroskopischer Hämaturie and Erythrozyturie
Eosinophilie in Bronchiallavage
Röntgen-Thorax
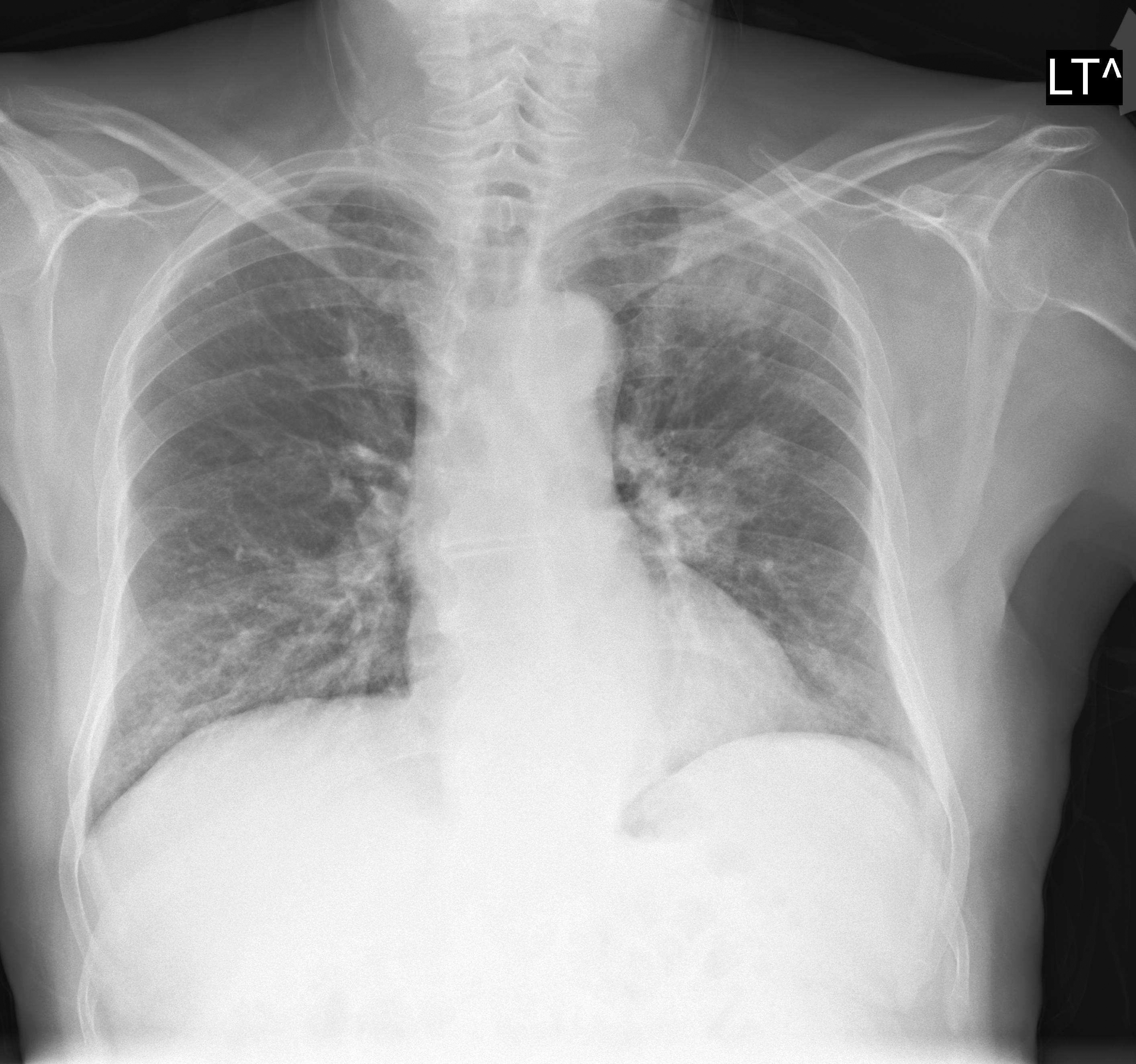
Bei 30 - 80% aller Fälle findet man Verschattungen der Lungen (Abb. 3), bei 25% ist das Röntgenbild unauffällig. Es kann multiple Verschattungen geben, die lokalisiert, bilateral, zentral oder peripher sein können.
Abb. 3: Röntgenbild mit nodulösen Verschattungen (linke Lunge) und infiltrativer Zeichnungsvermehrung (rechte Lunge)
Die Lungenverschattungen können flau sein und einer eosinophilen Pneumonie ähneln, sie können aber auch mehr nodulär sein. Auch Vergrößerungen und Verdichtungen der Hili sind gelegentlich möglich.
Bei pulmonaler alveolärer Kapillarbeteiligung sieht man gel. das Bild diffuser bilateraler alveolärer Infiltrate; in diesen Fällen treten in ca. 50 - 60% Hämoptysen auf.
Bei etwa ⅓ aller Patienten sieht man einen Pleuraerguß, der bei der mikroskopischen Untersuchung der Punktates eosinophil ist.
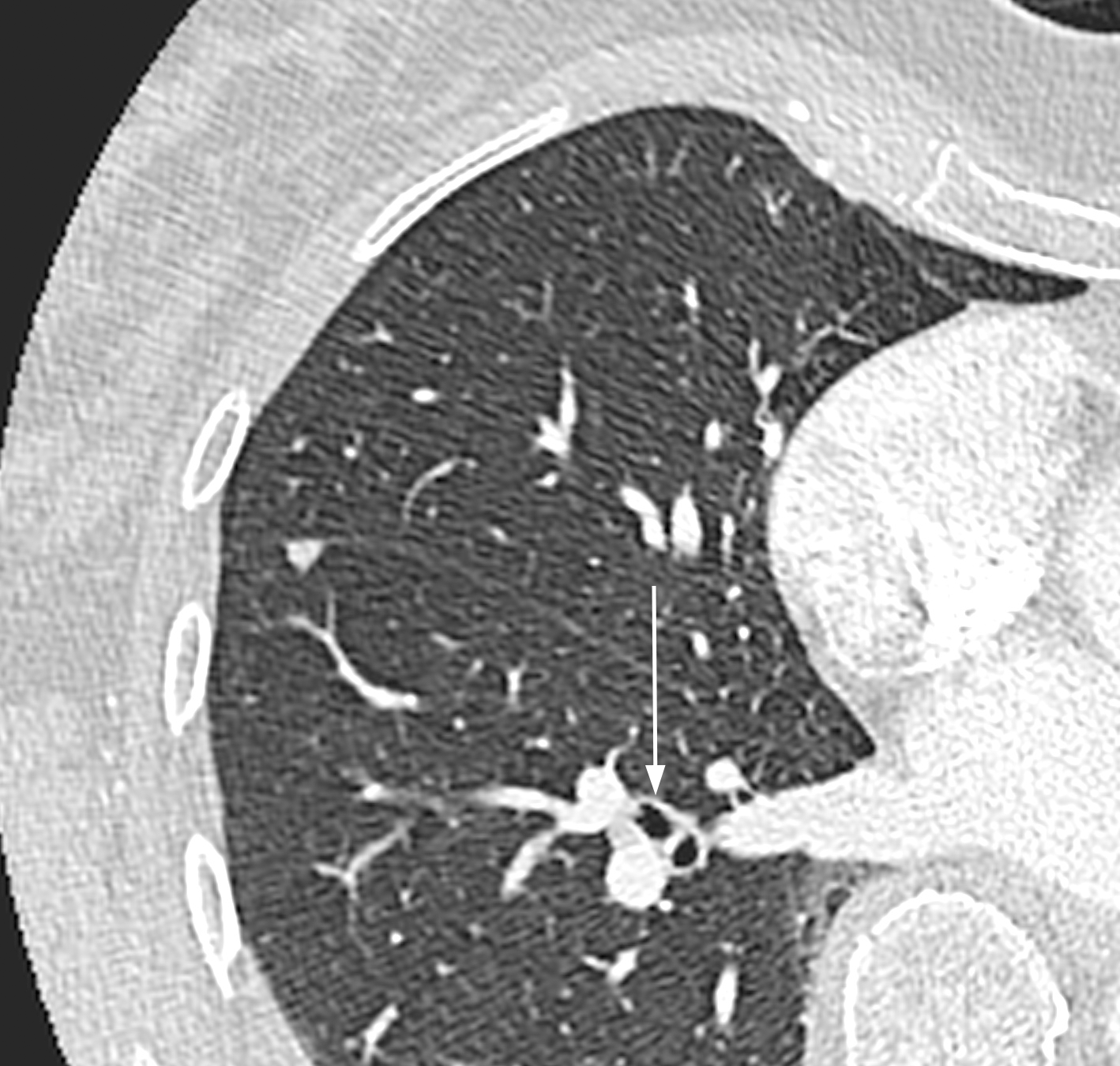
Thorax-CT
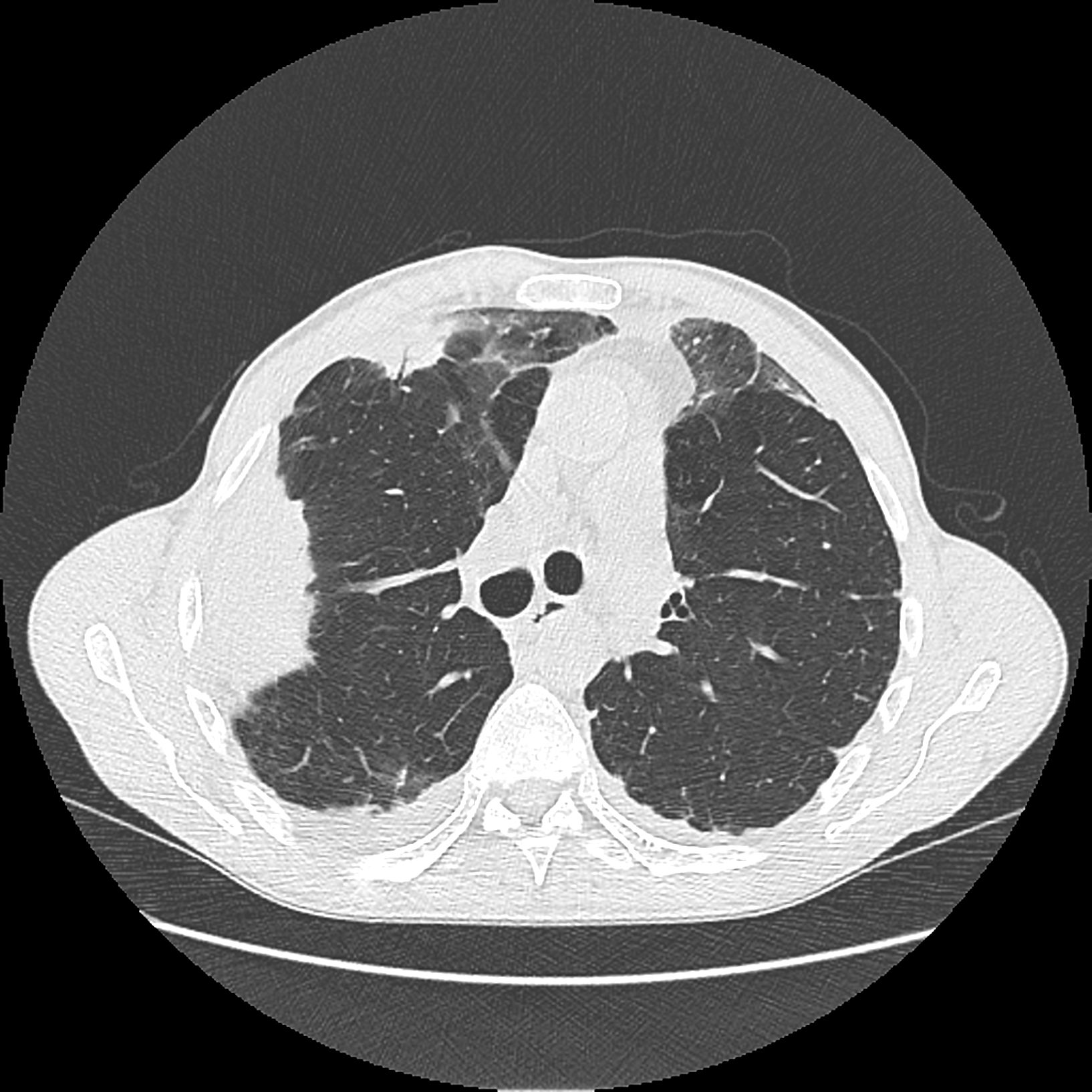
Ähnlich wie bei der eosinophilen Pneumonie sieht man uni- oder bilaterale milchglasartige Verschattungen (Abb. 4).
Abb. 4: Milchglasartige Verschattung in lateraler rechter Lunge
Manchmal, allerdings seltener, stellen sich auch noduläre Strukturen von 5 - 30 mm, die ein Cavum oder ein positives Bronchogramm zeigen können (Abb. 5).
Abb. 5: Noduläre Infiltration mit luftgefülltem Cavum
Auch Erweiterungen der Bronchien und deren Wandverdickung kann dargestellt werden.
In hochauflösenden CT-Bildern sieht man auch eine z.Z. erhebliche Erweiterung der peripheren Lungenarterien mit sternförmiger und unregelmäßiger Kontur als Zeichen einer Vaskulitis.
Andere Untersuchungen
Je nachdem welches Organ betroffen ist sieht man
im abdominellen CT den Befund einer Pankreatitis,
im EKG die Zeichen der myokardialen Ischämie, des Infarktes oder supraventrikuläre und ventrikuläre Arrhythmien,
im Echokardiogramm die Vergrößerung und diffuse Kontraktionsstörung des linken Ventrikels bei Myokarditis (Film 1),
Film 1: 4-Kammerblick bei diffuser Kontraktionsstörung des linken Ventrikels
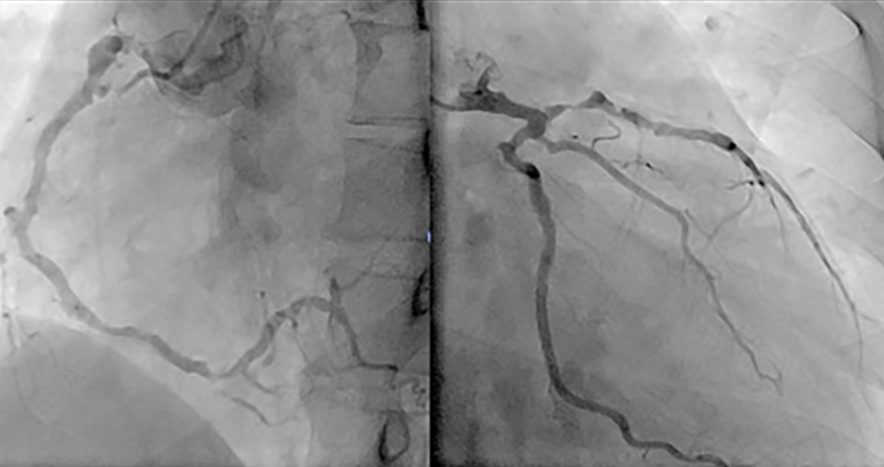
in der Coronarographie die Stenosen und Verschlüsse der Koronararterien bei myokardialer Ischämie oder Myokardinfarkt.
Abb. 3: Röntgenbild mit nodulösen Verschattungen (linke Lunge) und infiltrativer Zeichnungsvermehrung (rechte Lunge)
Beachten Sie die multiplen Stenosen und Wandunregelmäßigkeiten in der rechten (links) und der linken Koronararterie
Das Problem bei der coronarographischen Darstellung der Vasculitis ist, daß der Befund demjenigen einer arteriosklerotischen Koronarerkrankung vollkommen entspricht. Erst der klinische Kontext (z.B. junger Mensch, keine bekannten bedeutsamen arteriellen Risikofaktoren) lenkt den Verdacht in Richtung auf eine andere Ursache der Koronarveränderungen als eine Arteriosklerose. In diesen Fällen ist eine Laboruntersuchung (hohe Entzündungsparameter oder Eosinophilie) wegweisend und sollten dann auch Anlaßt für weitere Untersuchungen sein.
Wenn ein Koronarverschluß mit dem Bild eines akuten Koronarsyndroms auftritt wird man den Verschluß natürlich sofort rekanalisieren. Im Anschluß hieran muß bei der Suche nach der Ursache aber auch eine nicht-arteriosklerotische Ursache (CHURG-STRAUSS-Syndrom oder andere Vasculitiden) gedacht und in dieser Richtung weiter untersucht werden.
Wenn ein CHURG-STRAUSS-Syndrom bereits aus der Anamnese bekannt ist darf die Ursache des koronaren Problems eigentlich als geklärt angesehen werden.
Bei fortgeschrittenen Fällen einer koronaren Vaskulitis im Sinne einer schweren koronaren Mehrgefäßerkrankung stellt sich die Frage nach einer aortokoronaren Bypass-OP. Ich habe hierzu keine Literatur gefunden und kann nur eine eigene Casuistik beitragen:
Ein 38 Jahre alter Mann stellte sich mit den klinischen Beschwerden einer Koronarerkrankung vor. Bei der anschließenden Coronarographie fand sich eine schwere diffuse Koronarerkrankung mit zahlreichen, z.T. hochgradigen Stenosen in rechter und linker Koronararterie, ohne daß hierfür Risikofaktoren gefunden werden konnten.
Ich habe diesen Patienten zur Bypass-OP angemeldet, aber der Chirurg war ein erfahrener Mann, der dies zunächst ablehnte, weil er eine „entzündliche Ursache“ vermutete.
Bei der weiteren diagnostischen Aufarbeitung bestätigte letztlich ein Rheumatologe die Verdachtsdiagnose einer koronaren Vasculitis, ohne daß er eine konkrete Diagnose stellen konnte.
Der Patient wurde über mehrere Wochen hochdosiert mit Steroiden behandelt und nach 4 Monaten erneut coronarographiert.
Dabei fand sich eine deutliche Rückbildung der diffusen Koronarveränderungen mit jedoch dennoch verbliebenen hochgradigen Stenosen. Diese wurden dann operiert, der Patient erhielt 4 Bypass-Gefäße.
Er blieb weiter beim Kardiologen und Rheumatologen unter Kontrolle. Die Entzündungsparameter blieben niedrig, dem Patienten ging es kardial gut und ich habe ihn ca. 22 Jahre später aus den Augen verloren.
Kardio-CT und Kardio-MRT: Auch diese Techniken kann man einsetzen, um auf nicht invasive Weise den Zustand der Koronararterien und des linken Ventrikels zu untersuchen und um mittels MRT und einer Gadolinium-Gabe nach umschriebenen oder diffusen myokardialen Vernarbungen zu suchen,
bei der Gastroduodeno- bzw. Coloskopie die gastrointestinale Blutung und evtl. ihre Quelle,
im Elektromyogramm bzw. bei der Bestimmung der Nervenleitgeschwindigkeit die Zeichen der peripheren Neuropathie,
im MRT des Gehirns kann man im Ausbreitungsgebiet einzelner Gefäße hämorrhagische oder ischämische Infarkt sehen.
Biopsie
Sehr hilfreich, wenn nicht gar beweisend für die Klärung der Diagnose sind Biopsien aus dem betroffenen Organ.
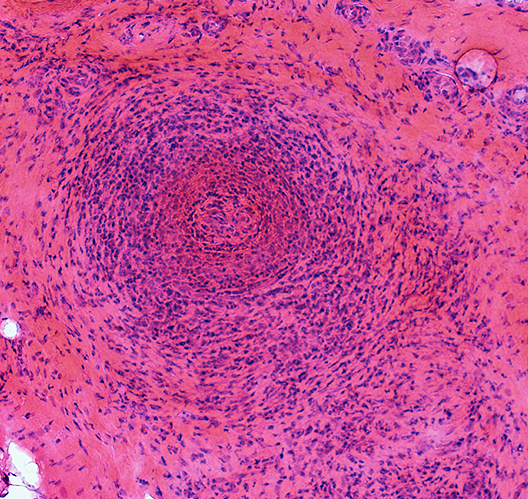
Hier sieht man den Befund einer Vasculitis mit eosinophilen Infiltraten der Gefäßwand und der Gefäßumgebung, oft verbunden mit dem Verlust des Gefäßlumens (Abb. 7). Diesen Gefäßbefund kann man wiederkehrend in allen Biopsien finden, die man aus den befallenen Organen entnimmt.
Abb. 7: Eosinophile Vasculitis einer kleinen Arterie
Beachten Sie die starke Vermehrung der Gefäßumgebung mit eosinophilen Zellen
In Abhängigkeit von den betroffenen Organen werden sie aber oft noch um organspezifische Befunde ergänzt, z.B.:
Bei Befall der Nieren sieht man bioptisch eine fokale oder rasch progrediente Glomerulonephritis, wenngleich ein solcher Befund nicht ein das CHURG-STRAUSS-Syndrom beweisender Befund ist, weil der mikroskopische Befund nicht von anderen Glomerulonephritiden unterschieden werden kann.
An der Lunge sieht man kleine nekrotisierende Granulome und (wie auch in anderen befallenen Organen) eine nekrotisierende Vaskulitis der kleinen Arterien und Venen.
Wenn es keine offenkundigen Zeichen eines Organbefalls gibt kann manchmal auch eine Muskel- oder Nervenbiopsie (N. suralis) helfen.
Therapie
Wie bei allen Autoimmunerkrankung ist eine kausale Therapie nicht möglich. Es wird somit eine medikamentöse Therapie erforderlich sein, deren Ziel es ist, das weitere Fortschreiten der Erkrankung aufzuhalten oder zu verhindern und die Beschwerden der Patienten zu lindern.
Weil die Erkrankung verschiedene Organe betreffen kann muß sie in gewisser Hinsicht und soweit als möglich organspezifisch ausgerichtet werden. Daher muß vor Einleitung einer Therapie festgestellt werden, welche Organe in welchem Schweregrad betroffen sind.
Man unterscheidet die Behandlung bei leichten, mittelschweren und schweren Erkrankungen. Dabei sind die schweren Fälle durch eine Beteiligung von Niere, Herz, Lungen (hämorrhagische Komplikationen) und Nerven mit den entsprechenden Funktionsstörungen gekennzeichnet.
Grundlage der Therapie ist die Gabe von Steroiden im Sinne einer Monotherapie. Sie können, auch in relativ niedriger Dosis das CHURG-STRAUSS-Syndrom gut kontrollieren. Üblicherweise tritt hierunter nach etwa 4 Wochen eine Besserung ein.
Steroide bewirken eine Verminderung der Eosinophilie und eine Rückbildung der Gewebsinfiltrate.
Leichte und mittelschwere Fälle
Die Behandlung beginnt mit der oralen Gabe von 0.5 - 1.0 mg Prednisolon/kg KG täglich, wobei man eines Tagesdosis von 60 mg nicht überschreiten sollte.
In der Regel kommt es beim CHURG-STRAUSS-Syndrom hierdurch innerhalb weniger Tage zu einer Rückbildung der Eosinophilie und Leukozytose, sowie zum Absinken von BSG und C-reaktivem Protein. Tritt diese Verbesserung nicht zügig innerhalb weniger Tage ein muß man dies als einen Hinweis auf eine ungünstige Prognose ansehen.
Bei gutem Ansprechen auf diese Therapie setzt man sie zunächst für etwa 3 Wochen fort. Danach reduziert man die Dosis um 5 mg alle 10 Tage bis zu einer Dosis von 0.5 mg/kg/d. Diese Dosis wird über die folgenden 10 Tage um 2.5 mg tgl. reduziert, bis man das Steroid nach insgesamt etwa 3 Monaten absetzen kann.
Die Rückfallrate mit dieser Therapie liegt bei etwa 25%. In diesen Fällen erfolgt eine Behandlung wie unten für die schweren Fälle beschrieben wird.
In Fällen, bei denen eine Steroid-Monotherapie nicht innerhalb weniger Tage ausreicht oder in denen sich die Erkrankung in einem schon fortgeschrittenen Stadium mit Befall von Herz, Nieren, Lungen oder Nerven befinden wird die Steroidtherapie verändert und zusätzlich ein Immunsuppressivum geben, wie dies für die schweren Fälle beschrieben wird. Eine solche therapeutische Eskalation ist bei etwa 20% der Patienten notwendig.
Schwere Fälle
In diesen Fällen erfolgt eine Kombinationstherapie mit einem Immunsuppressivum:
Die primäre Steroid-Therapie besteht nicht in der Gabe von Prednisolon, sondern von Methylprednisolon, das parenteral gegeben wird.
Man beginnt mit 15 mg/kg KG intravenös in Kombination mit dem oralen Prednison (1 mg/kg KG) über zunächst 3 Tage. Danach beendet man die parenterale Gabe des Methylprednisolons und schließt eine orale Prednisolon-Therapie mit 1 mg/kg KG oral an. Dabei sollte aber eine Tagesdosis von 80 mg/d nicht überschreiten werden.
Zusätzlich setzt man ein Immunsuppressivum ein. Hierzu verwendet man Cyclophosphamid, das als wiederholte hochdosierte Kurzinfusion („pulsed therapy“) für einen Zeitraum von 3 - 6 Monaten gegeben wird. Eine solche „gepulste“ Behandlung ist dazu gedacht, das Risiko der Nebenwirkungen einer oralen Cyclophosphamid-Therapie zu vermindern, die Effizienz dieser gepulsten Therapie ist aber noch nicht sicher erwiesen.
Die orale Cyclophosphamid-Therapie, deren Dosis u.a. nach der Leukozytenzahl im Blut gesteuert wird sollte 6-12 Monate nach Erreichen der Remissionsphase fortgesetzt werden.
Die Kombination von Steroid und oraler Cyclophosphamid-Therapie verbessert den Zustand der Patienten und hilft zudem bei der Einsparung der Steroid-Dosis. Ungefähr 2 Wochen nach Beginn der Therapie kann man versuchen, die Steriod-Dosis zu reduzieren.
Spezielle Aspekte
In vielen mittelschweren und schweren Fällen verhindert die Persistenz des Asthma bronchiale eine Verminderung der Prednisolon-Dosis auf weniger als 10 - 15 mg/d. Hier ist oft eine langfristige Gabe von Steroiden erforderlich.
Bei einigen Patienten, die unter steroidaler Monotherapie schwer zu behandeln sind kann man die orale oder parenterale Steroidtherapie mit einer Plasmaaustausch-Behandlung oder Plasmapherese kombinieren.
Auch kann man erwägen, eine intravenöse Behandlung mit Immunglobulin durchzuführen wie bei der WEGENER´schen Granulomatose oder der mikroskopischen Polyangiitis. Diese Behandlung kann in einigen Fällen zur klinischen Verbesserung und zur Verminderung des ANCA-Titers im Serum führt.
Auch in Kombination mit Steroiden kann man das Antirheumatikum Dapson einsetzen. Vor allem bei CHIRG-STRAUSS-assoziierter Myokarditis ist es bewiesenermaßen wirksam. Wenn es unter dieser Therapie zu einer Verbesserung der Myokarditis kommt kann man die Dosis des Steroids reduzieren.
Kardiomyopathie: Unter der Therapie mit Steroiden und Cyclophosphamid sprechen eine Myokarditis ebenso wie die schweren Formen einer systemischen Vaskulitis nur schlecht an. Bei Myokarditiden mit nur geringer Einschränkung der linksventrikulären Pumpfunktion kann eine Steroidtherapie zu einer gewissen Verbesserung der systolischen und diastolischen Ventrikelfunktion führen, bei stark eingeschränkter Pumpfunktion ist dies allerdings nicht zu erwarten. In diesen Fällen kann man versuchen, die Pumpfunktion durch die Gabe von Immunglobulinen zu verbessern.
Neuropathie: Wenn es unter der initialen Steriodtherapie zu einer Verbesserung der Symptomatik kommt darf man erwarten, daß dieser Erfolg über mehrere Monate auch nach Beendigung der Therapie anhält. Tritt diese Verbesserung jedoch nicht ein kann der zusätzliche Einsatz von Cyclophosphamid und/oder einer Immunglobulingabe helfen.
Die medikamentöse Behandlung des CHURG-STRAUSS-Syndroms ist nichts, was ein Kardiologe (egal ob in Praxis oder Klinik) alleine durchführen sollte. Sie sollte vielmehr primär durch einen Rheumatologen erfolgen. In Abhängigkeit von den betroffenen Organen muß die Behandlung aber oft interdisziplinär sein und neben einem Kardiologen und einem Pneumologen auch Nephrologen, Gastroenterologen und sogar einen Onkologen involvieren.
Nachsorge
Angesichts des Charakters der Erkrankung und des Umstandes, daß die die Gabe der Immunsuppressiva eine Langzeit-Therapie ist müssen die Patienten regelmäßig und engmaschig durch einen Rheumatologen und ggfs. die anderen Fachgebiete kontrolliert werden.
Bei den Verlaufskontrollen werden u.a. der klinische Zustand des Patienten, Eosinophilenzahl im Blut und die laborchemischen Entzündungsparameter beobachtet, während die Höhe des ANCA-Titers nicht gut mit dem Aktivitätsgrad der Erkrankung korreliert.