Was ist das QT-Syndrom?
Das QT-Syndrom (heutige Bezeichnung: Long-QT-Syndrom (LQTS)) ist eine seltene Krankheit, die bei sonst herzgesunden Menschen zum plötzlichen Herztod führen kann. Es ist entweder vererbt oder im Laufe des Lebens erworben, meist als Folge einer unerwünschten Arzneimittelwirkung. Man unterscheidet die QT-Zeit-Verlängerung von einem QT-Syndrom (siehe „Beschreibung der Erkrankung“).
Erklärung
 |
| Abb.1 |
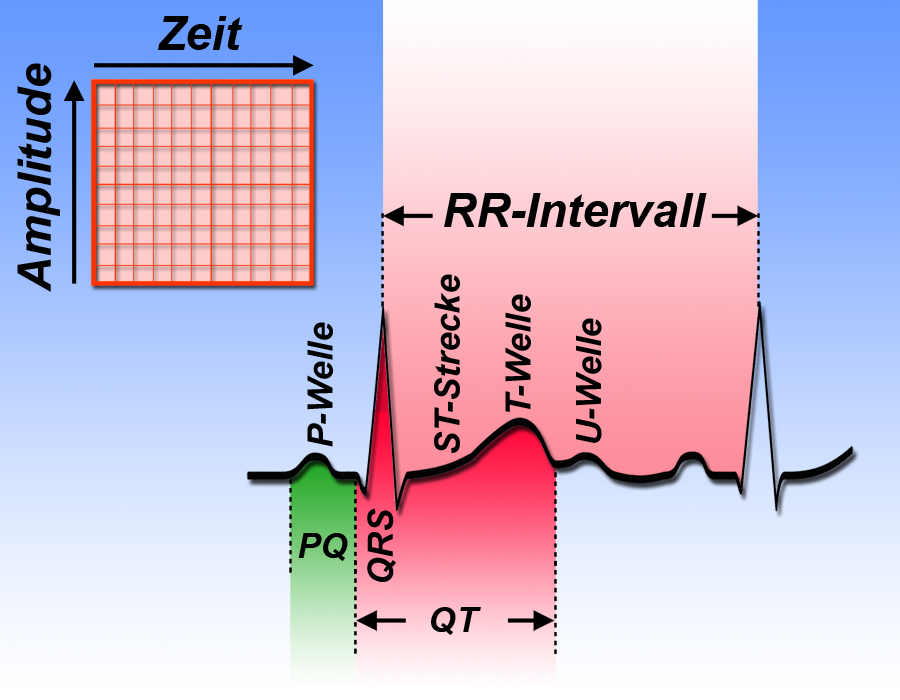
QT bezieht sich auf das Zeitintervall zwischen dem Beginn der Q-Zacke und dem Ende der T-Welle im EKG (Abb. 1).
Dieser Zeitabschnitt zeigt die sog. „Erregungsrückbildung“ des Herzens an. Wenn Sie Genaueres wissen möchten klicken Sie hier; an dieser Stelle nur soviel:
Ein Herzschlag entsteht dadurch, daß die Herzmuskelzellen elektrisch erregt werden und sich hierdurch zusammen ziehen. Nach jeder solchen elektrischen Erregung muß sich der Herzmuskel wieder entspannen, um bereit für die nächste Erregung und damit für den nächsten Herzschlag zu sein.
Diese Periode im elektrischen Ablauf eines Herzschlages bezeichnet man als „Erregungsrückbildung“.
Erregung und Erregungsrückbildung einer Zelle (auch einer Herzmuskelzelle) entstehen durch bestimmte Salze (= Elektrolyte, vor allem Natrium, Kalium, Kalzium und Magnesium), die elektrisch geladen sind (elektrisch geladene Teilchen = Ionen (sprich: Johnen)).
 |
| Abb.2 |
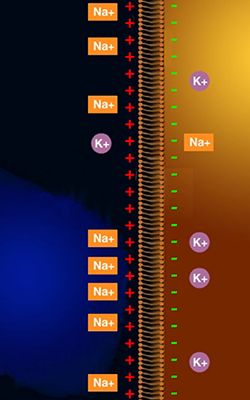
Die Konzentrationen dieser Ionen sind innerhalb und außerhalb einer Zelle unterschiedlich, sodaß an der äußeren Zellwand eine elektrische Spannung entsteht (weil sich außerhalb der Zelle mehr positiv geladene (Natrium-) Salze befinden als innerhalb).
Sie sehen diese unterschiedliche Verteilung der Elektrolyte im Ruhezustand der Zelle in Abb. 2.
Sie sehen, daß außerhalb der Zelle (links im Bild) mehr positiv geladene Natrium-Ionen als innerhalb der Zelle und daß innerhalb der Zelle mehr positiv geladene Kalium-Ionen liegen als außerhalb. Außerhalb der Zelle befinden sich somit netto mehr positiv geladene Ionen als innerhalb, daher ist das Zelläußere positiv gegenüber dem Zellinneren geladen.
Wenn sich nun eine Muskelzelle z.B. des Armes zusammenziehen soll dann bekommt sie den Befehl hierzu über einen Nerven (am Herzen erfolgt dies mehr oder weniger automatisch; Sie werden dies verstehen, wenn Sie die Broschüre oder das eBook über den Aufbau und die Funktion des Herzens und hier das eBook über die Anatomie mit der „elektrischen System des Herzens“ lesen.)
Wenn sich ein Muskel also zusammenziehen soll dann wird die Zellwand schlagartig für bestimmte Elektrolyte (Natrium) durchlässig.
Die Folge ist, daß große Mengen Natrium von außerhalb der Zelle in deren Inneres strömen.
Weil das Natrium elektrisch positiv geladen ist verursacht dies nun eine schlagartige Veränderung der elektrischen Spannung der Zellwand, weil nun nämlich (anders als im Ruhezustand der Zelle) eine große Menge positiv geladenen Natriums in die Zelle eingedrungen ist, was dazu führt, daß nun das Innere der Zelle gegenüber ihrem Äußeren positiv geladen ist. Diese schlagartige Spannungsänderung führt nun innerhalb der Zelle zu Vorgängen, an deren Ende das Zusammenziehen der Muskelzelle steht.
Nach einen solchen Zusammenziehen des Muskels (= Kontraktion) muß die normale Verteilung der Elektrolyte wiederhergestellt werden, damit sich der Muskel wieder „elektrisch erholen“ kann und bereit für seine nächste Kontraktion ist.
Diese Bereitschaft zu einer erneuten Erregung der Muskelzelle setzt voraus, daß die Verteilung der Elektrolyte innerhalb und außerhalb der Muskelzelle wieder „normalisiert“ wird.
Hierzu muß das in die Zelle eingedrungene Natrium schnell wieder aus der Zelle entfernt werden und auch die Konzentrationen anderer Blutsalze, die im Rahmen der elektrischen Erregung das Zellinnere verlassen haben (z.B. positiv geladenes Kalium) wieder in den Normalzustand versetzt werden. Zuständig hierfür sind sogenannte „Natrium-Kalium-Pumpen“, sowie bestimmte Transportkanäle (Ionenkanäle, z.B. langsamer oder schneller Natriumkanal), die nur ganz spezielle Elektrolyte transportieren und die in der Zellwand eingebaut sind.
| Film 1 |
Durch die Tätigkeit dieser Pumpen wird das normale Konzentrationsgefälle der einzelnen Elektrolyte und damit die normale Spannung der Zellwand schon nach sehr kurzer Zeit wieder hergestellt und die Zelle ist für ihre nächste Aktion bereit (Film 1).
 |
| Abb.3 |
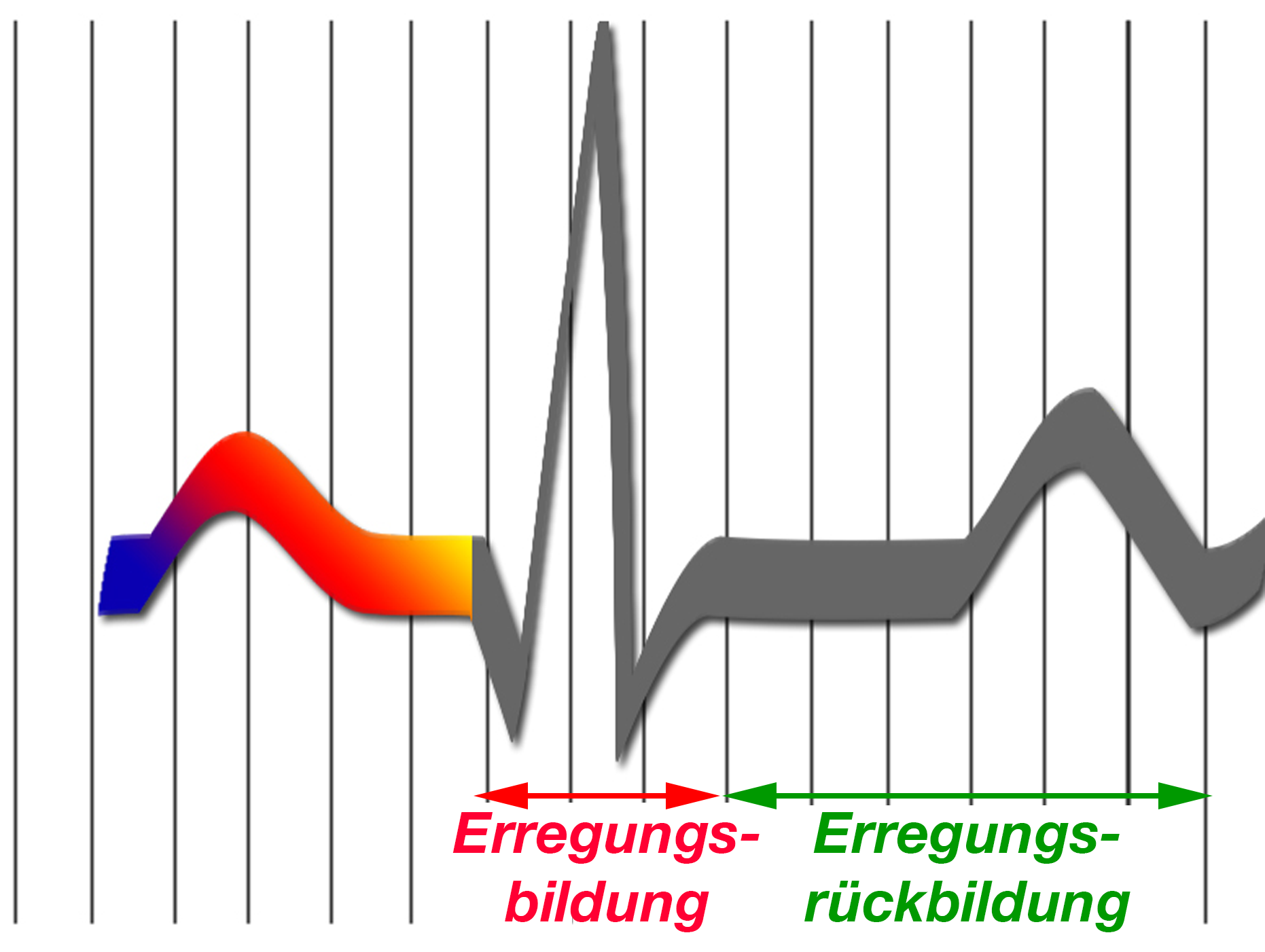
Erregungs- und die Erregungsrückbildungsphase kann man im EKG erkennen (Abb. 3).
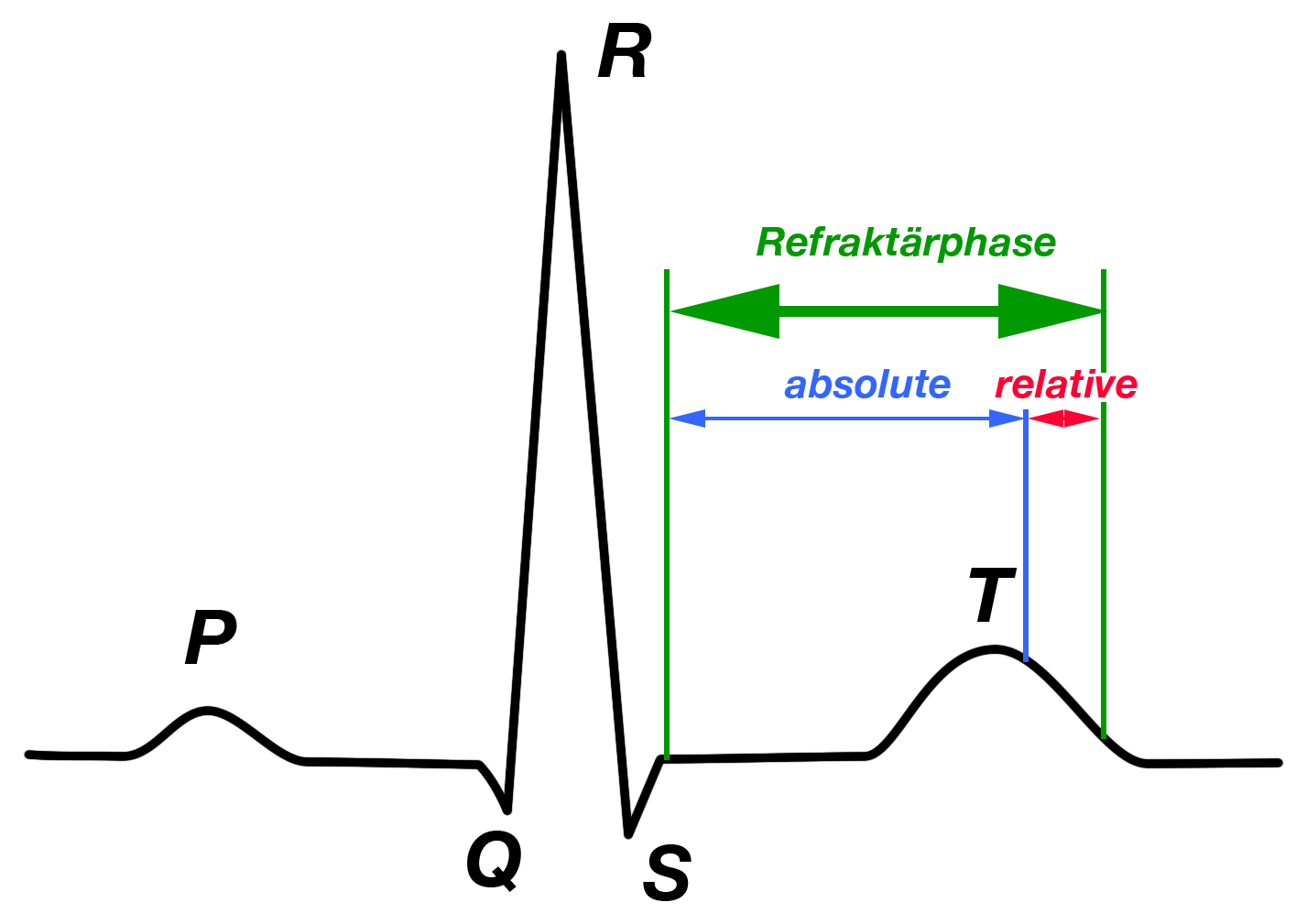
Zu Beginn der Erregungsrückbildungsphase ist die Zelle nicht erneut erregbar, unabhängig davon, wie und womit man diese Zelle erneut zu einer Aktivität anregen möchte; man nennt diese Phase „absolute Refraktärperiode“.
 |
| Abb.4 |
Im Anschluß daran folgt eine kurze Phase, in der die Zelle nur durch einen überdurchschnittlich starken Reiz wieder zu einer erneuten Aktivität veranlaßt werden kann („relative Refraktärperiode“) (Abb. 4).
Diese Zeitabläufe verlaufen in der Gesamtheit aller Herzmuskelzellen nicht absolut synchron, d.h. es gibt Phasen, in denen einige Zellen noch absolut refraktär sind und daneben andere Zellen, die bereits relativ refraktär sind.
| Film 2 |
| Normale Erregungsausbreitung (Zeitlupe) |
Im Normalfall ist diese Zeit der Phase aber nur sehr kurz (Film 2).
Wenn in der Herzmuskelwand in einem solchen Fall mit nahezu synchron ablaufenden Refraktärzeiten ein Extraschlag („Extrasystole“) auftritt dann breitet sich die elektrische Erregung vom Ort der Entstehung des Extraschlages wellenförmig, d.i. gleichförmig aus (Film 3).
| Film 3 |
Bei einem QT-Syndrom ist die Erregungsrückbildung jedoch gestört, indem die Transportkanäle (z.B. Natrium-Kalium-Pumpe) in den Zellmembranen für Natrium, Kalium und Eisen nicht mehr richtig funktionieren.
Die Folge ist eine Verlängerung oder Verkürzung der absoluten und relativen Refraktärzeit.
Die Störung der Transportkanäle ist bei Menschen mit dem QT-Syndrom von Zelle zu Zelle sehr unterschiedlich ausgeprägt. So kann es geschehen, daß unmittelbar neben einer absolut unerregbaren Zelle (absolute Refraktärzeit) auch andere Zellen liegen, die noch „etwas unerregbar“ (relative Refraktärzeit) oder sogar schon wieder vollkommen normal erregbar sind. Sehen Sie dies in Film 4 und in Abb. 5.
 |
|
| Film 4 | Abb. 5 |
| Ausschnitt aus der Erregungsrückbildungsphase des Herzmuskels. | |
| Beachten Sie die verschiedenen Stadien der Erregungsrückbildung in den einzelnen Zellen (grün = normal erregbare Zelle, verschiedene Rottöne = Zellen in unterschiedlichen Stadien der Erregungsrückbildung) |
Man nennt dies „Inhomogenität der Erregungsrückbildung“.
Wenn nun ein elektrischer Extraschlag auftritt dann hängen dessen Auswirkungen davon ab, zu welchem Zeitpunkt der Erregungsrückbildung er auftritt:
| Film 5 |
Es kann geschehen, daß der elektrische Impuls zu einem Zeitpunkt auftritt, zu dem eine Kette bereits wieder erregbarer Zellen direkt neben noch unerregbaren Zellen liegt.
In diesen Fällen können sogenannte kreisende Erregungen entstehen (Film 5):
Der zusätzliche Reiz erregt eine einzelne Zelle, die bereits relativ refraktär und damit erregbar ist. Diese Zelle gibt die Erregung an denjenigen ihrer Nachbarn weiter, der bereits ebenfalls relativ refraktär geworden ist, während die anderen Zellen, die sich noch in der absoluten Refraktärperiode befinden nicht reagieren.
Aus diesem kreisenden Erregung“ können dann die umgebenden Muskelzellen aktiviert werden, wie dies in Film 5 mit den Pfeilen symbolisiert wird.
Hierdurch entsteht eine bösartige Form des Herzrasens, die u.U. tödlich enden kann (siehe unter „Krankheitserscheinungen“).
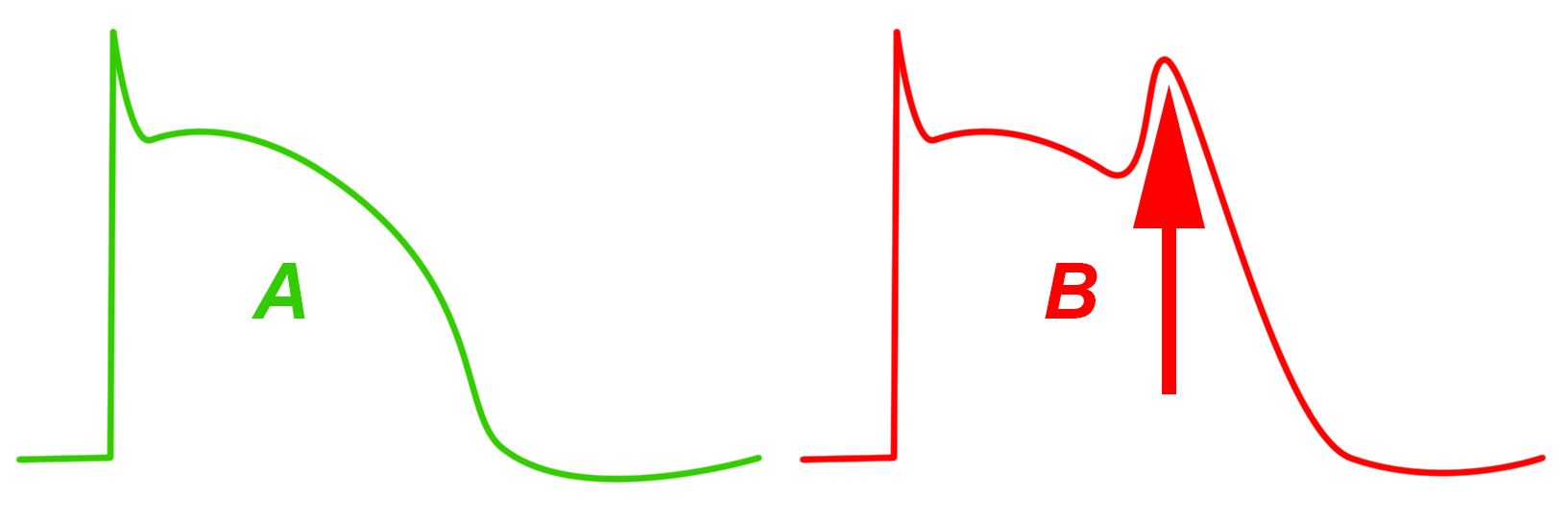 |
| Abb.6 |
| Links (grün) Ablauf einer normalen elektrischen Erregung (Membranpotential) einer Muskelzelle der Herzkammer. Rechts (rot) Membranpotential mit „Nachdepolarisation“ (Pfeil) |
Beim QT-Syndrom kommt aber noch etwas hinzu:
Ebenfalls durch die fehlerhaft arbeitenden Elektrolyt-Transportkanäle können sog. „Nachdepolarisationen“ auftreten. Dabei handelt es sich um Spannungsschwankungen während der elektrischen Rückbildungsphase der Zelle (Abb. 6).
Solche Nachdepolarisationen können ebenfalls zu abnormen Erregungen der anderen Herzmuskelzellen führen.
Entweder (und auch dies ist abhängig vom Zeitpunkt der Nachdepolarisation) können „kreisende Erregungen“ wie oben beschrieben auftreten; es kann aber auch dazu kommen, daß irgendwelche in der Kammerwand gelegenen Muskelzellen erregt und damit aktiv werden.
| Film 6 |
| Der blaue Impuls symbolisiert den Ursprung des Kammerflimmerns |
Und weil diese Zellen irgendwo in der Herzwand liegen und weil sie nach dem Zufallsprinzip erregt werden werden unterschiedliche Muskelzellen zu unterschiedlichen Zeitpunkten elektrisch erregt. Es findet in diesen Fällen keine geordnete und aufeinander abgestimmte Tätigkeit der einzelnen Herzmuskelzellen auf, sondern es kommt zur Anarchie (Film 6):
Viele Muskelzellen arbeiten völlig unkoordiniert und heben sich in ihrer Wirkung gegenseitig auf. Das Herz bleibt mechanisch stehen, das Herz kann kein Blut mehr pumpen, der Kreislauf bleibt stehen und der Mensch stirbt. Dieses Ereignis nennt man Kammerflimmern.
Das QT-Syndrom ist daher deshalb so gefährlich, weil aus banalen Herzstolperschlägen oder manchmal auch ohne solche Extrasystolen lebensgefährlich und sogar tödlich Herzrhythmusstörungen entstehen können.
Beschreibung der Erkrankung
Einteilung
Man unterscheidet 2 Formen einer QT-Zeit-Verlängerung:
- Reine Verlängerung der QT-Zeit („QT-Zeit-Verlängerung“)
- long-QT-Syndrom.
Auch hier unterscheidet man wieder 2 Formen:
- Angeborenes long-QT-Syndrom
- Erworbenes long-QT-Syndrom
Reine QT-Zeit-Verlängerung
Ein „Syndrom“ ist definiert als die Kombination von Befunden und Symptomen, die typischerweise gemeinsam auftreten. Bezüglich der QT-Zeit sind solche Befunde bzw. Symptome:
- Verlängerung der QT-Zeit
- Schwindel
- Ohnmacht
- (überlebter) plötzlicher Herztod
- Schwerhörigkeit
Besteht lediglich eine Verlängerung der QT-Zeit im EKG, weist der Betroffene aber keine weiteren Symptome aus der oben stehenden Liste auf so spricht man von einer reinen QT-Zeit-Verlängerung.
Ist die Verlängerung der QT-Zeit hingegen mit einem oder mehreren Symptomen aus der oben stehenden Liste kombiniert spricht man vom QT-Syndrom.
QT-Syndrome
Angeborene Formen
Hier unterscheidet man 2 Krankheiten:
Das Romano-Ward- und das Jervell-Lange-Nielsen-Syndrom.
Beiden Syndromen gemeinsam ist die Verlängerung der QT-Zeit (siehe unter EKG).
Beim Romano-Ward-Syndrom tritt zusätzlich aber noch eine Schwerhörigkeit auf, Kinder mit Jervell-Lang-Nielsen-Syndrom hören normal.
Die Störung der Erregungsrückbildung wird in allen angeborenen Fällen durch Störungen von Erbanlagen (= Genen) verursacht, die für die Herstellung von Ionenkanälen verantwortlich sind.
Erworbene Formen
Eine Verlängerung der QT-Zeit kann durch
- eine Vielzahl von Medikamenten
- durch sog. Elektrolytstörungen (z.B. bei Nierenerkrankungen, Diätfehler)
- bei Entzündungen des Herzmuskels (Myokarditis) oder
- bei Durchblutungsstörungen des Herzmuskels (Koronarerkrankung mit oder ohne Herzinfarkt)
auftreten.
Medikamente
Zunächst bekannt wurde ein Zusammenhang zwischen einem long-QT-Syndrom und Medikamenten bei bestimmten Herzrhythmusmedikamenten (= Antiarrhythmika).
Dies bezieht sich auf Medikamente, die Chinidin und den ß-Blocker Sotalol enthalten. Mittlerweile hat man aber mehrere hundert Medikamente gefunden, die die QT-Zeit u.U. gefährlich verlängern können.
Hierzu gehören neben Chinidin und Sotalol z.B.:
- Antibiotika wie Erythromycin und Trimethoprim-Sulfamethoxazol
- einige Antihistaminika
- viele Psychopharmaka, wie beispielsweise Neuroleptika
- Parkinsonmittel
- Anti-Malaria-Medikamente
- Röntgenkontrastmittel oder
- verschiedene Opioide (Schmerzmittel) incl. Methadon.
Mehrere Präparate sind deswegen bereits vom Markt genommen worden.
Eine Liste dieser Medikamente, die immer wieder auf den neuesten Stand gebracht wird finden Sie im Internet, wenn Sie hier klicken.
Es gibt andere Listen, bei denen Sie sich mit Ihrer eMail-Adresse anmelden müssen. Dies ist praktisch, damit man automatisch via eMail über Veränderungen (z.B. neue als gefährlich angesehene Medikamente) informiert werden kann.
Gemeinsam ist diesen Substanzen, daß sie in der Herzmuskelzelle den Ausstrom des Kalium-Ions während der Erregungsrückbildung hemmen und dadurch das QT-Intervall verlängern können. Das Risiko für derartige unerwünschte Arzneimittelwirkungen ist
- bei niedrigen Pulsfrequenzen
- Frauen
- erniedrigtem Kaliumspiegel im Blut
- Verdickung des Herzmuskels durch die Bluthochdruckkrankheit
- bei Herzmuskelschwäche und
- hohen Wirkstoffkonzentrationen dieser Medikamente
erhöht.
Krankheitserscheinungen
Eine Verlängerung der QT-Zeit spürt man normalerweise nicht, mehr als die Hälfte der Patienten mit einem Long-QT-Syndrom leiden an keinerlei Beschwerden.
Treten dennoch Symptome auf, so handelt es sich um
- Herzrasen
- Schwindel oder
- plötzlicher Bewußtlosigkeit (= Synkope).
Verursacht werden diese Symptome durch das Auftreten bestimmter Herzrhythmusstörungen (anhaltende oder nicht-anhaltende ventrikuläre Tachykardie, Kammerflimmern oder Torsade de points) (siehe „Notfälle“), die je nach Art und Dauer der Herzrhythmusstörung zum Herzstillstand und damit zum Tod führen können.
Die Herzrhythmusstörung tritt urplötzlich und bevorzugt bei körperlicher Belastung oder in Stresssituationen unerwartet und aus völligem Wohlbefinden heraus auf. wenn man von Sportlern hört, die plötzlich auf dem Spielfeld zusammengebrochen sind und dann entweder wiederbelebt werden mußten oder die sogar gestorben sind handelt es sich meistens um Menschen mit einem bis dahin unbekannten long-QT-Syndrom.
Häufigkeit und Prognose
Plötzliche Todesfälle junger und ansonsten gesunder Menschen sind statistisch gesehen selten und noch seltener sind sie Folge eines long-QT-Syndroms. Man nimmt an, daß in Deutschland jährlich etwa 10 - 20 Menschen unter 30 Jahre an einem long-QT-Syndrom sterben, die Zahl beruht aber auf Schätzungen.
Angeborene Long-QT-Syndrome treten mit einer Häufigkeit von 1:5.000 bis 1:15.000 aller Lebendgeburten auf. Etwa 30-40% dieser Patienten erleiden vor dem 40. Lebensjahr eine Synkope. Die Häufigkeit eines plötzlichen Herztodes bei Heranwachsenden im Alter von 10-20 Jahren mit long-QT-Syndrom liegt bei ca. 1,5%.
Bestimmte Patienten, die man mittels eines normalen EKGs und einer molekulargenetischen Untersuchung findet werden als Hochrisikopatienten angesehen. Sie haben ohne Behandlung ein Risiko von mehr als 50%, vor ihrem 40. Lebensjahr eine Synkope, einen Herzstillstand oder den plötzlichen Herztod zu erleiden.
Untersuchungsmöglichkeiten
|
| Abb.7 |
Der wegweisende und namensgebende Befund des long-QT-Syndroms ist die Verlängerung des QT-Intervalls im Ruhe-EKG (Abb. 7).
Die in Millisekunden gemessene QT-Zeit ist für sich genommen wenig aussagekräftig, da sie beim Menschen u. a. von der Herzfrequenz, dem Alter und dem Geschlecht abhängig ist.
Um eine abnormal lange QT-Zeit zuverlässig erkennen und verschiedene QT-Zeiten im Verlauf miteinander sinnvoll vergleichen zu können, ist eine rechnerische Korrektur der gemessenen QT-Zeit erforderlich, wobei hierzu am häufigsten die Bazett-Formel genutzt (QTc ist die korrigierte QT-Zeit):
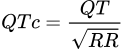 |
(QTc = korrigierte QT-Zeit, QT = QT-Zeit aus EKG, RR = Abstand 2er R-Zacken). (Moderne EKG-Geräte berechnen die „korrigierte QT-Zeit automatisch.)
Als oberer Grenzwert gilt eine QTc-Zeit von 440 ms, ab 500 ms ist von einem hohen Risiko auszugehen.
st die QTc-Zeit verlängert bedeutet dies (siehe oben) ein gewisses Risiko für die Entstehung bösartiger Herzrhythmusstörungen, man kann aber noch nicht zwingend von einem long-QT-Syndrom sprechen.
Um diese Diagnose stellen zu können kann der Kardiologe einen Score benutzen, in den nicht nur die Dauer der QTc-Zeit einfließen, sondern auch andere EKG-Befunde, bestimmte Ereignisse aus der Vorgeschichte (z.B. Ohnmachtszustände), der körperlichen Untersuchung (z.B. Schwerhörigkeit) oder der Krankheitsgeschichte der Familie (z.B. plötzlicher Herztod bei Familienangehörigen unter 30 Jahren oder das bekannte Vorliegen eines long-QT-Syndroms bei einem Blutsverwandten).
Bei jungen Menschen und immer dann, wenn keine Medikamente eingenommen werden, die die QT-Zeit verlängern können, ist es darüber hinaus sinnvoll, eine molekulargenetische Untersuchung durchzuführen. Eine solche Untersuchung klärt zwar die Diagnose des long-QT-Syndroms nicht, sagt aber viel über das individuelle Risiko des betroffenen Menschen aus.
Bei den angeborenen Formen des long-QT-Syndroms sollten ..........
Ende der Leseprobe
- Krankheiten mit ähnlichen Erscheinungen
- Komplikationen
- Notfälle
- Vorbeugende Maßnahmen
- Verhaltensweisen, die die Heilung fördern
- Verhaltensweisen, die die Krankheit verschliummern
- Therapie
- Wann muß der Hausarzt kontaktiert werden?
- Ein Anhang mit einer Ausstellung zahlreicher Medikamente, die bei Menschen mit QT-Syndrom gefährlich sind
Die Informationen auf dieser Seite finden Sie in Band 18a einer eBook-Reihe der Patienten-Akademie.
Hier bekommen Sie dieses eBook in verschiedenen Formaten:
- padBook (für iPad und epub3-fähige eBook-Reader)
- phoneBook (für smartPhones)
- Paperwhite (für Kindle Paperwhite)